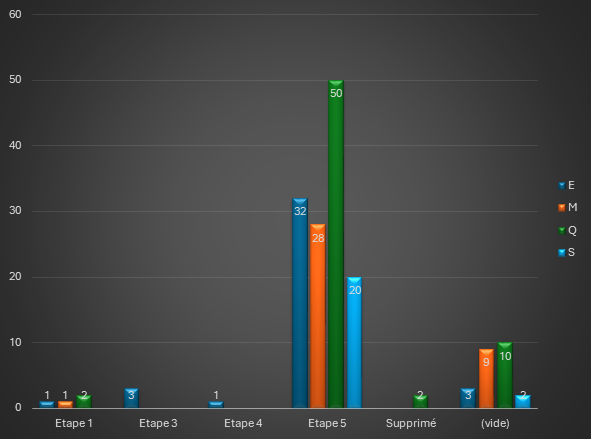
Quelles différences entre une Procédure de Reconnaissance Mutuelle et une Procédure Décentralisée ?
D’un point de vue législatif, la procédure de reconnaissance mutuelle est définie par la directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 établissant un code communautaire relatif aux médicaments à usage humain.
Par la suite, c’est la directive 2004/27/CE qui a prévu les bases de la procédure décentralisée.
Ces deux procédures sont accessibles à toutes les demandes d’autorisation de mise sur le marché qui n’entrent pas dans le champ d’application obligatoire de la procédure centralisée. Elles sont applicables dès lors qu’un demandeur souhaite enregistrer son médicament dans plus d’un Etat membre.
1/ La Procédure de Reconnaissance Mutuelle (MRP)
L’article 28.2 de la directive 2001/83/CE telle que modifiée, spécifie le champ d’application de cette procédure : le principe de cette procédure est d’étendre une AMM nationale,déjà obtenue dans un des Etats de l’Union Européenne, à un ou plusieurs autres Etat(s) membre(s) (ou « CMS » pour Concerned Member State(s)) dans lesquels le laboratoire souhaite commercialiser son médicament. L’Etat référent (ou « RMS » pour « Reference Member State »), qui a octroyé l’AMM existante, pilote la procédure.
Après clôture de la procédure d’évaluation, les AMM sont délivrées par chacune des autorités compétentes des Etats membres concernés.
Source: EMA
Après une éventuelle mise à niveau du dossier d’AMM,le RMS envoie aux CMS le dossier d’AMM et son rapport d’évaluation incluant, le résumé des caractéristiques du produit (RCP), la notice et l’étiquetage, 14 jours avant le démarrage de la procédure.
Les Etats Membres concernés devront alors reconnaître l’autorisation déjà délivrée par le RMS dans un délai de 90 jours, sans arrêt de l’horloge (ou « clock-stop »)).
La procédure peut toutefois se terminer au jour 60 si les CMS n’ont plus de commentaires.
Une fois que l’AMM (y compris le RCP, la notice et l’étiquetage) est reconnue par les CMS, cette reconnaissance est suivie d’une phase nationale de clôture de 30 jours prévue pour délivrer l’AMM nationale. Les procédures closes avec succès sont publiées sur le site du CMDh.
Si un Etat membre a des objections pour reconnaître le rapport d’évaluation relatif au dossier, le résumé des caractéristiques du produit (RCP), la notice et l’étiquetage, en raison d’un risque potentiellement grave pour la santé publique (comme défini par l’article 29(1) de la directive 2001/83/CE), le dossier est renvoyé au CMDh pour une discussion approfondie. Cette procédure dure 60 jours. Si le CMDh est dans l’incapacitié de trancher à l’issue de ces 60 jours, la demande est envoyée au CHMP pour arbitrage (article 29(4) de la directive 2001/83/CE). Cette procédure dure également 60 jours.
2/ La Procédure Décentralisée (DCP)
Cette procédure se distingue de la MRP par deux différences principales :
- Aucune AMM ne doit avoir été préalablement accordée dans l’UE,
- Le dossier est soumis simultanément dans tous les Etats membres.
Dans ce cas, le laboratoire demande à un État membre d’agir en tant qu’État de référence (« RMS ») afin de réaliser l’évaluation, parmi les États au sein desquels il souhaite obtenir une autorisation pour son médicament.
Source EMA
* EEA = European Economic Area
MA = Marketing authorization
Le RMS établit un rapport d’évaluation préliminaire relatif au dossier soumis et aux projets de RCP, de notice et d’étiquetage.
Ce rapport est soumis aux CMS et au demandeur au jour 70 de la procédure pour commentaires.
Au jour 105 de la procédure, l’horloge s’arrête pour permettre au demandeur de soumettre des éléments de réponse aux questions soulevées par les Etats membres à l’issue de la 1ère phase.
Lorsque les réponses ont été soumises, l’horloge repart au jour 106 et, au jour 120 de la procédure, le RMS fait circuler, en parallèle, une mise à jour de tous les documents (rapport d’évaluation, RCP, notice et étiquetage) au demandeur et aux CMS.
Une 2ème phase de questions/réponses débute et la procédure peut se clôturer au jour 150 si tous les commentaires ont été résolus.
Autrement, une nouvelle phase de 60 jours démarre afin de finaliser les questions en suspens.
La procédure DCP dure donc au maximum 210 jours (hors arrêts d’horloge). Cette période est suivie d’une phase nationale de clôture de 30 jours prévue pour délivrer les AMM nationales. Les procédures closes avec succès sont publiées sur le site du CMDh.
Comme pour la procédure MRP, s’il n’y a pas de consensus entre les Etats membres, le dossier est renvoyé au CMDh pour une discussion approfondie. Cette procédure dure 60 jours. Si le CMDh est dans l’incapacitié de trancher à l’issue de ces 60 jours, la demande est envoyée au CHMP pour arbitrage (article 29(4) de la directive 2001/83/CE). Cette procédure dure également 60 jours.
3/ En résumé :
| MRP | DCP |
| AMM initiale nationale existante | Pas d’AMM accordée dans l’UE |
| Pas de choix du RMS (AMM nationale déjà existante dans l’UE) | Le choix du RMS relève du demandeur |
| Demande une reconnaissance par les autres Etats membres | Demande simultanée dans tous les Etats membres : le RMS évalue le dossier pour la première fois (comme les CMS) |
| Seulement une phase d’évaluation | Deux phases d’évaluation |
| Décision en 90 jours, pouvant aller jusqu’à 150 jours en cas d’arbitrage par le CMDh si pas de consensus entre les Etats membres | Décision en 210 jours (hors arrêt d’horloge), pouvant aller jusqu’à 270 jours en cas d’arbitrage par le CMDh si pas de consensus entre les Etats membres |
| Dossier d’AMM identique dans tous les Etats Membres | |
| Principe de reconnaissance de l’évaluation de l’État membre de Référence (RMS) par les autres États membres concernés (CMS) | |
| Le choix des Etats impliqués dans ces procédures relève du demandeur | |
| Phase nationale de clôture de 30 jours prévue pour délivrer l’AMM nationale | |
| Un rapport européen public d’évaluation (ou Public Assessment Report « PAR ») pour chaque médicament approuvé via DCP/MRP est publié dans le répertoire « Mutual Recognition Index » par le RMS sur le site de la HMA | |
ATESSIA accompagne les laboratoires dans tout le processus de l’enregistrement : de la stratégie d’enregistrement à la rédaction et soumission des dossiers d’AMM.
Article rédigé par Mathilde ISRAEL, Consultante en Affaires Réglementaires et Communication Externe.