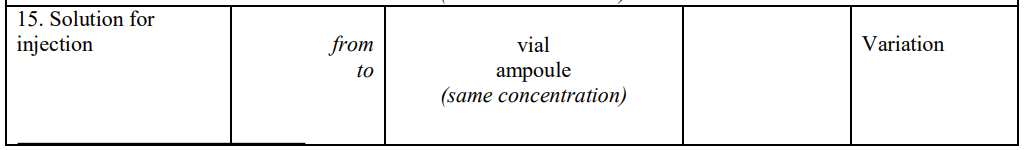
ATESSIA, Votre Guide en Affaires Réglementaires pour l’Industrie Pharmaceutique
Dans le monde complexe et hautement réglementé de la pharmacie et des soins de santé, ATESSIA se distingue comme un cabinet de conseil dédié à accompagner les entreprises dans les défis des affaires réglementaires. Consciente du rôle essentiel de la conformité et de la sécurité des patients, ATESSIA propose une expertise complète couvrant la documentation scientifique, les essais cliniques, les soumissions réglementaires et la pharmacovigilance. En assurant une collaboration fluide avec les autorités de santé dans plusieurs juridictions, ATESSIA aide les entreprises à atteindre la conformité tout en favorisant leur croissance, avec un engagement constant pour la sécurité des patients et l’efficacité des produits.
Les Affaires Réglementaires : Un Moteur de Croissance
Les affaires réglementaires ne sont pas qu’une série de tâches administratives ; elles constituent un véritable levier stratégique de croissance et d’innovation dans les secteurs pharmaceutique et des dispositifs médicaux. Elles garantissent avant tout que les produits respectent les normes de sécurité et de qualité les plus élevées, depuis leur développement jusqu’à leur commercialisation. Cela implique l’alignement des processus de fabrication sur les exigences légales, la communication continue avec les autorités sanitaires, et la gestion rigoureuse de la documentation requise pour les soumissions réglementaires.
Les affaires réglementaires jouent également un rôle clé de coordination, en favorisant la collaboration entre les équipes cliniques, médicales et marketing pour garantir que toutes les étapes du développement répondent aux normes réglementaires. Au-delà de la conformité, elles soutiennent la croissance stratégique en guidant les efforts de recherche et développement, en accélérant l’accès au marché via les cadres réglementaires, et en facilitant les partenariats et accords de licence. Enfin, l’expertise réglementaire permet aux entreprises d’accéder efficacement à de nouveaux marchés, tout en maintenant la conformité des produits existants face à l’évolution des réglementations.
Trouver un équilibre dans la charge réglementaire
Une charge réglementaire excessive peut épuiser les ressources d’une entreprise et freiner son innovation. Les solutions proposées par ATESSIA répondent à ce défi en allégeant les pressions internes, permettant ainsi aux équipes de se concentrer sur des activités clés telles que les essais cliniques et la stratégie commerciale.
En collaborant avec ATESSIA, les entreprises bénéficient d’une approche équilibrée des affaires réglementaires. L’expertise d’ATESSIA accélère l’accès au marché en gérant efficacement les soumissions réglementaires et garantit des opérations à la fois rentables et maîtrisées. En tant que cabinet indépendant, ATESSIA offre des conseils objectifs et impartiaux, permettant aux clients de naviguer en toute confiance dans les complexités du paysage réglementaire.
Ce qui rend les Affaires Réglementaires françaises uniques et comment ATESSIA peut vous aider
Le marché pharmaceutique français présente des défis et opportunités spécifiques liés à son cadre réglementaire unique. Les exigences strictes de l’ANSM (Agence Nationale de Sécurité du Médicament et des Produits de Santé), associées à des lois spécifiques comme le Code de la Santé Publique, créent un environnement complexe pour assurer la conformité. ATESSIA agit comme un partenaire stratégique pour les entreprises internationales, leur offrant des conseils experts pour naviguer dans ces spécificités et prospérer sur le marché français.
Les défis majeurs incluent l’obtention du statut d’Exploitant, le respect des exigences en matière d’étiquetage et de conditionnement, la mise en place de systèmes de pharmacovigilance robustes, ainsi que la conformité aux régulations publicitaires et à la Loi Anti-Cadeau. ATESSIA accompagne également ses clients dans la négociation des prix et des remboursements avec la HAS (Haute Autorité de Santé) et le CEPS (Comité Économique des Produits de Santé), garantissant des stratégies d’entrée sur le marché optimales et conformes.
L’Engagement d’ATESSIA en matière d’expérience client
Sous la direction de Géraldine Baudot-Visser, ATESSIA repose sur des principes de précision, de flexibilité et une compréhension approfondie des besoins de ses clients. L’entreprise privilégie des solutions sur-mesure, élaborant des stratégies réglementaires alignées sur le contexte, le secteur et les objectifs stratégiques de chaque projet. La transparence est au cœur de l’approche d’ATESSIA, avec une communication claire et continue qui permet aux clients de rester informés et confiants à chaque étape.
Cette vision de partenariat à long terme positionne ATESSIA comme bien plus qu’un simple consultant réglementaire. En s’adaptant aux évolutions du paysage réglementaire et en soutenant ses clients dans leur croissance, ATESSIA devient un allié essentiel pour atteindre la conformité et favoriser une croissance durable.
ATESSIA transforme les affaires réglementaires, d’un défi opérationnel en un outil stratégique pour le succès. En offrant un soutien et une expertise incomparables, l’entreprise aide les acteurs du secteur pharmaceutique à surmonter les défis réglementaires, à garantir leur compétitivité et à saisir les opportunités de croissance.
Pour plus d’informations, rendez-vous sur www.atessia.fr ou contactez-nous à hello@atessia.fr. L’équipe d’ATESSIA est prête à optimiser vos affaires réglementaires et à soutenir vos objectifs stratégiques.
Pour voir l’article complet sur Silicon Review, c’est ici !