Quelles sont les différentes options pour l’enregistrement d’une substance active dans un dossier d’Autorisation de Mise sur le Marché (AMM) en Europe ?
La description des données relatives à la substance active fait partie des éléments obligatoires devant figurer dans le Module 3 du dossier d’AMM. Ces éléments doivent être présentés selon le plan général décrit dans la Directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain.
Toutefois, si le fond des données attendues est précisé dans la réglementation européenne, la matérialisation de ces dernières dans le dossier d’AMM peut différer, selon l’option sélectionnée par le demandeur lors de l’agrément de son fabricant de substance active.
En Europe, l’enregistrement d’une substance inscrite par le demandeur d’AMM peut se faire via un ASMF/DPSA1 (cf. Directive 2001/83/EC, Annexe 1, Partie 1, Chapitre 3.2, Article (8)) ou via un CEP (cf. Directive 2001/83/EC, Annexe 1, Partie 1, Chapitre 3.2, Article (7)), le choix de la procédure étant laissé au fabricant de la substance active (aucune n’étant obligatoire).
Une autre possibilité est d’envisager la soumission, par le demandeur dans son dossier d’AMM, des données relatives à la substance active sous la forme d’une documentation scientifique dite « complète ».
On distingue différents cas de figures selon le type de substance active :
Type de substance active :
Voie documentaire applicable :
Nouvelle entité chimique (NtA, Volume 2A, Chapitre 1, Annexe I)
ASMF / DPSA ou documentation complète
Substance inscrite à la Pharmacopée européenne
ASMF / DPSA ou CEP ou documentation complète
Substance non-inscrite à la Pharmacopée européenne
ASMF / DPSA ou documentation complète
Dans cet article, nous nous attachons à expliciter les principales différences entre le CEP et l’ASMF pour enregistrer une substance active dans une autorisation de mise sur le marché en Europe.
Focus sur la procédure de CEP
Pour des informations complémentaires sur le CEP, consultez notre article de Blog « Qu’est-ce que les CEP ? ».
Focus sur la procédure d’ASMF / DPSA
Les requis pour la constitution d’un ASMF / DPSA en Europe sont décrits dans le guide de l’EMA intitulé « Final Guideline on Active Substance Master File Procedure (CHMP/QWP/227/02, EMEA/CVMP/134/02) » et le document de questions-réponses associé intitulé « Q&A on Active Substance master file (ASMF) » conjoint à l’EMA et au CMDh.
L’objectif principal de la procédure relative à l’ASMF est de permettre la protection de la propriété intellectuelle ou du « savoir-faire » confidentiel du fabricant de la substance active, tout en permettant au demandeur ou au titulaire de l’AMM d’assumer l’entière responsabilité du médicament, tout en garantissant la qualité et le contrôle de la qualité de la substance active.
Les autorités nationales compétentes / l’EMA ont ainsi accès à toutes les informations nécessaires pour évaluer l’adéquation de l’utilisation de la substance active dans le médicament.
Résumé des différences entre l’utilisation d’un CEP et d’un ASMF / DPSA
Un bref comparatif des avantages/inconvénients des procédures de CEP et d’ASMF / DPSA pour documenter la partie relative à la substance active dans un dossier d’AMM est présenté ci-après.
Critère
CEP
ASMF / DPSA
Confidentialité des données du fabricant de substance active
Très forte L’ensemble du Module 3.2.S est confidentiel (partagé avec l’EDQM uniquement, pas avec le TAMM)
Forte Module 3.2.S divisé en deux parties : > Partie fermée confidentielle > Partie ouverte non-confidentielle
Évaluation
Module 3.2.S évalué par EDQM (évaluation mutualisée) à Questions adressées uniquement au fabricant de la substance active
Parties ouverte et fermée évaluées par l’autorité où l’ASMF est déposé (évaluation non-mutualisée – sauf en cas de procédure de répartition des tâches « ASMF worksharing ») à Questions sur la partie fermée directement adressées au fabricant de la substance active ; questions sur la partie ouverte adressées au TAMM
Charge de travail pour le TAMM
Faible
Moyenne
Charge de travail pour le fabricant de substance active
Élevée au départ, faible ensuite
Moyenne
Gestion des variations d’AMM en lien avec la substance active
Très efficace : variations via des mises à jour du CEP
Moyenne
Quand est-ce recommandé ?
Substance avec monographie Ph. Eur. / commercialisation étendue de la substance active
Substance avec ou sans monographie Ph. Eur. / souhait de protection du savoir-faire
Les procédures réglementaires, les délais d’évaluation et les coûts associés sont différents.
Les consultants d’ATESSIA vous accompagnent dans la rédaction de vos dossiers de CEP, d’ASMF / DPSA et leur soumission auprès des autorités.
- EUR-Lex – Directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain
– EudraLex – Volume 2 – Notice to Applicants, Volume 2A, Chapter 1, Annex I
L’innovation dans le domaine du médicament est complexe, coûteuse et fortement réglementée. Pour accompagner les porteurs de projets innovants, les start-ups, PME, industriels ou chercheurs, l’Agence européenne du médicament (EMA) a mis en place plusieurs dispositifs d’accompagnement scientifique et réglementaire.
Contrairement à d’autres organismes, l’EMA ne finance pas directement les projets, mais elle joue un rôle clé pour sécuriser et accélérer le développement des innovations médicamenteuses.
Le rôle de l’EMA dans l’innovation
L’EMA est responsable de l’évaluation et de la supervision des médicaments au niveau européen. Son objectif est double :
garantir la sécurité, la qualité et l’efficacité des médicaments centralisés,
favoriser l’accès rapide à l’innovation, notamment lorsque des besoins médicaux ne sont pas couverts.
Pour cela, elle propose des outils permettant d’interagir très tôt avec les développeurs afin d’anticiper les enjeux réglementaires.
1. Innovation Task Force (ITF) : le point d’entrée pour les projets innovants
L’Innovation Task Force (ITF) est un groupe multidisciplinaire de l’EMA qui offre une plateforme de discussion sur les enjeux scientifiques, réglementaires et légaux des médicaments innovants, comme les thérapies avancées (ATMPs).
Les réunions d’orientation ITF permettent aux développeurs, qu’ils soient chercheurs académiques, consortiums européens, PME ou grandes entreprises, d’engager un dialogue précoce et informel avec l’EMA. Ces échanges, gratuits et organisés en ligne, aident à clarifier les questions techniques et scientifiques avant même le conseil scientifique formel.
L’ITF apporte un soutien particulier aux domaines clés de l’innovation, tels que l’intelligence artificielle, les technologies plateformes, les nouvelles approches méthodologiques et les principes des 3Rs pour la recherche animale. En facilitant ces échanges précoces, l’ITF aide les projets innovants à réduire l’incertitude réglementaire et à mieux planifier leur développement.
2. Le SME Office :un soutien stratégique pour les PME
Le SME Office de l’EMA accompagne les petites et moyennes entreprises dans le développement et la commercialisation de médicaments au sein de l’Union européenne et de l’Espace économique européen. Pour bénéficier de ce soutien, les entreprises doivent demander le statut PME et répondre aux critères européens correspondants.
Une fois le statut accordé, les PME peuvent profiter de réductions ou d’exonérations sur les frais liés aux procédures réglementaires, comme les avis scientifiques, les inspections et la pharmacovigilance. Elles bénéficient également de services pratiques supplémentaires, comme la traduction gratuite de la documentation produit dans toutes les langues de l’UE pour les premières autorisations de mise sur le marché, ce qui simplifie grandement les démarches administratives.
De plus, les PME sont inscrites dans le registre public des PME de l’EMA, conçu pour faciliter les interactions, partenariats et collaborations avec d’autres acteurs du secteur.
3. Scientific Advice et Protocol Assistance : sécuriser la stratégie de développement
Le Scientific Advice de l’EMA aide les développeurs à s’assurer que leurs tests et études sont appropriés et bien conçus, réduisant ainsi les risques que des objections majeures soient soulevées lors de l’évaluation d’une demande d’autorisation de mise sur le marché. Cette démarche protège également les patients en évitant qu’ils participent à des études qui ne produiraient pas de données utiles.
L’avis scientifique est fourni par le CHMP (Committee for Medicinal Products for Human Use), sur recommandation du Scientific Advice Working Party (SAWP). Pour les médicaments destinés à traiter, prévenir ou diagnostiquer des maladies à risque pour la santé publique (comme la COVID‑19 ou la grippe aviaire), le CHMP se base sur les recommandations de la Emergency Task Force (ETF).
Le Protocol Assistance couvre aussi les aspects des demandes d’essais cliniques, permettant aux développeurs de sécuriser toutes les étapes réglementaires et scientifiques avant le lancement des études cliniques. En pratique, ce service réduit les risques réglementaires et facilite la planification du développement, en particulier pour les médicaments innovants ou à haute urgence sanitaire.
4. Qualification des nouvelles méthodologies : reconnaître les approches innovantes
La qualification des nouvelles méthodologies permet à l’EMA d’évaluer et de donner un avis officiel sur des méthodes ou outils innovants utilisés dans le développement de médicaments. Cela peut concerner des biomarqueurs, des méthodes d’imagerie, des modèles expérimentaux ou toute approche scientifique appliquée aux études non cliniques ou cliniques.
Le processus est supervisé par le CHMP, qui se base sur les recommandations du Scientific Advice Working Party (SAWP). L’évaluation repose sur les données soumises par le développeur et inclut souvent une consultation publique de la communauté scientifique, garantissant transparence et discussion ouverte.
Dans certains cas, avant que la méthodologie ne soit pleinement qualifiée, l’EMA peut émettre une lettre de soutien. Ces lettres résument la méthodologie, son contexte d’utilisation et les données disponibles, et encouragent le partage de données et la poursuite d’études pour aboutir à la qualification officielle.
En résumé, ce dispositif permet de valider des méthodes innovantes avant leur adoption généralisée, offrant aux développeurs une reconnaissance réglementaire et une plus grande sécurité scientifique pour leurs projets.
5. Quality Innovation Group (QIG): accompagner l’innovation dès la fabrication
Le Quality Innovation Group (QIG) réunit des experts en qualité des médicaments chimiques et biologiques, y compris les produits de thérapie avancée (ATMPs), ainsi que des spécialistes des inspections de bonnes pratiques de fabrication. Ce groupe a été créé pour aider les développeurs dès les premières étapes du cycle de vie d’un produit, en facilitant les discussions avec différents acteurs : experts ad hoc, universitaires, évaluateurs et inspecteurs européens, régulateurs internationaux, associations industrielles et académiques, ainsi que via le EU Innovation Network.
Le QIG soutient l’utilisation de méthodologies innovantes dans le développement des médicaments, telles que des technologies nouvelles, des matériaux ou dispositifs innovants, ou encore la digitalisation des procédés de fabrication. Son objectif est de fournir un avis cohérent tout au long du développement du produit, que ce soit lors de procédures réglementaires formelles (scientific advice, protocol assistance, demandes d’autorisation de mise sur le marché ou procédures post‑autorisation) ou lors de réunions informelles permettant de repérer tôt les questions réglementaires potentielles.
6. EU-Innovation Network (EU-IN) : une plateforme de collaboration pour l’innovation
L’EU Innovation Network (EU‑IN) est un groupe de travail dont l’objectif est de renforcer la collaboration entre l’EMA et les autorités nationales compétentes sur les aspects réglementaires liés aux thérapies et technologies émergentes. Ce groupe soutient également le développement des médicaments innovants et des technologies associées. Sa mission principale est de soutenir le développement de ces innovations, en comblant les lacunes dans le soutien réglementaire précoce, en offrant une plateforme pour l’échange des bonnes pratiques et en favorisant l’engagement direct avec les innovateurs, afin de mieux accompagner les projets innovants à travers l’Europe.
7. PRIME : un soutien prioritaire pour les médicaments à fort impact
Les avantages incluent :
un accompagnement réglementaire renforcé,
des interactions plus fréquentes avec l’EMA,
la possibilité d’une évaluation accélérée.
PRIME est particulièrement stratégique pour les innovations de rupture à fort impact clinique.
Le programme PRIME (PRIority MEdicines) est une initiative de l’EMA destinée à soutenir le développement des médicaments répondant à un besoin médical non satisfait. Il s’agit d’un dispositif volontaire qui repose sur un dialogue précoce et renforcé avec les développeurs, afin d’optimiser les plans de développement et d’accélérer l’évaluation, permettant ainsi aux patients d’accéder plus rapidement à des traitements prometteurs.
Grâce à PRIME, l’EMA fournit un soutien proactif pour la génération de données robustes sur les bénéfices et risques des médicaments, et facilite l’accélération de l’évaluation des demandes d’autorisation de mise sur le marché. Le programme s’appuie sur le cadre réglementaire et les outils existants, comme les avis scientifiques et l’évaluation accélérée. Les développeurs dont les médicaments bénéficient de PRIME peuvent s’attendre à une procédure d’évaluation accélérée au moment de la demande d’autorisation.
En favorisant un engagement précoce avec les développeurs, PRIME contribue à améliorer la qualité des preuves scientifiques et à s’assurer que les données collectées sont adaptées à l’évaluation réglementaire, tout en permettant aux patients de bénéficier plus rapidement de thérapies susceptibles d’améliorer significativement leur qualité de vie.
En résumé
Les aides à l’innovation de l’EMA reposent sur un principe clé : mieux accompagner pour mieux innover.
Même sans financement direct, ces dispositifs permettent de :
réduire les risques réglementaires,
accélérer le développement,
améliorer les chances d’accès au marché européen.
Pour toute entreprise ou équipe développant un médicament innovant, interagir tôt avec l’EMA est un véritable levier stratégique. Atessia accompagne ses clients tout au long de ces démarches, que ce soit pour les réunions d’orientation, le SME Office, le Scientific Advice ou les programmes comme PRIME, afin de sécuriser et d’optimiser le développement de leurs médicaments innovants.
Article rédigé par Lamya SAOUSSEN, Consultante Junior en Affaires Réglementaires et Communications Externes
Le code CIP (Code Identifiant de Présentation) est un identifiant numérique unique attribué à chaque présentation pharmaceutique autorisée en France. Il permet de distinguer précisément un médicament selon son nom, son dosage, sa forme pharmaceutique et son conditionnement et la contenance de son conditionnement.
Depuis le décret n° 2021‑1931 du 30 décembre 2021, le code CIP bénéficie d’une existence légale, rendant son attribution et son usage obligatoires pour l’ensemble des médicaments autorisés sur le marché français.
Attribué par l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) à l’issue de la procédure d’enregistrement ou d’autorisation de mise sur le marché (AMM), le code CIP est utilisé tout au long du circuit du médicament. Il facilite l’identification, la facturation, la gestion des stocks et la traçabilité sanitaire des produits de santé.
Les spécifications techniques ont été définies par l’arrêté du 30 décembre 2021 portant sur les modalités d’attribution et de codification des médicaments.
Le code CIP est distinct du code NL, associé à un dossier de demande d’AMM, et du code CIS, associé à un médicament.
Dans le circuit hospitalier, l’identification peut également reposer sur le code UCD (Unité Commune de Dispensation), qui permet d’identifier la plus petite unité de médicament dispensée à l’hôpital. Le code UCD est attribué par l’ANSM et ne peut exister que s’il est rattaché à un code CIP.
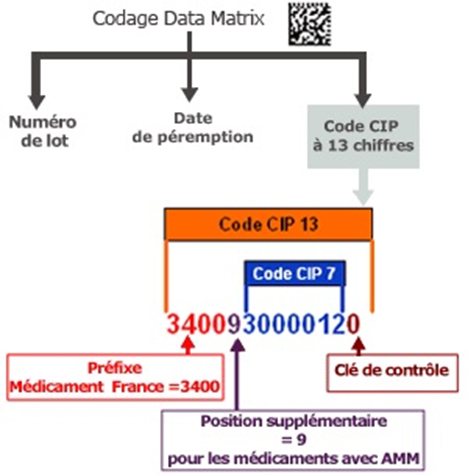
Comment est structuré le code CIP 13 ?
Depuis le 1er janvier 2009, le code CIP à 7 chiffres a été remplacé par un code à 13 chiffres, en raison de la saturation du format initial et de l’évolution du cadre réglementaire. Ce passage au CIP 13 permet notamment d’intégrer, dans un marquage lisible automatiquement, la date de péremption et le numéro de lot, conformément aux exigences renforcées en matière de sécurité sanitaire.
Le code CIP 13 est structuré de la manière suivante :
« 3400 » : préfixe indiquant le secteur du médicament en France ;
« 9 » : chiffre de rubrique désignant une présentation pharmaceutique ;
7 chiffres : code CIP 7, identifiant spécifique de la présentation du médicament (forme, dosage, conditionnement) ;
1 chiffre : clé de contrôle, calculée selon l’algorithme EAN‑13.
Ce code, lorsqu’il est précédé d’un « 0 », respecte les normes ISO/IEC 15459-3 2014 et ISO/IEC 15459-4 2014. Ce format est compatible avec les codes-barres lisibles automatiquement, tels que de type EAN-13 ou Data Matrix, apposés sur les conditionnements et garantissant une lecture fiable dans les pharmacies, établissements de santé, et logiciels métiers.
Dans le circuit hospitalier, le code UCD repose également sur un format à 13 caractères numériques. Les cinq premiers chiffres sont « 34008 » et le dernier chiffre correspond à une clé de contrôle calculée selon la formule de Luhn.
À quoi sert le code CIP ?
Le code CIP permet de :
Faciliter la gestion des stocks et des flux d’approvisionnement, ainsi que la constitution des marchés.
Participer à la lutte contre la falsification des médicaments.
Identifier précisément chaque spécialité pharmaceutique, avec suivi des autorisations de mise sur le marché et contrôle de conformité.
Traiter les demandes de remboursement, suivre les dépenses et codifier les tarifs.
Assurer la traçabilité, la pharmacovigilance et la transmission d’alertes liées aux effets indésirables ou aux rappels de lots.
Actuellement, une réflexion est en cours par le Club Inter Pharmaceutique (CIP) sur l’avenir de la codification des médicaments en France. La décision d’évoluer ou non vers le code VIP2400 reviendra au ministère de la santé.
Depuis 2018, le risque de contamination des médicaments par des impuretés de type nitrosamines s’est imposé comme l’un des sujets les plus sensibles en matière de qualité pharmaceutique. Ce qui n’était au départ qu’un signal isolé est devenu un véritable enjeu réglementaire mondial, mobilisant autorités, industriels et laboratoires de contrôle. Comprendre ce risque, ses origines et ses implications est désormais indispensable pour tout acteur du médicament.
1. D’où vient le risque nitrosamines ?
Les nitrosamines sont des composés classés comme potentiellement cancérogènes. Elles peuvent se former dans certaines conditions chimiques, notamment en présence :
d’un agent nitrosable : précurseur (ex. amine secondaire, tertiaires, quaternaire et sel d’ammonium),
d’un agent nitrosant (ex. nitrites),
d’un environnement favorable (pH acide, température élevée, phase aqueuse, impuretés).
Il existe 2 principaux types de nitrosamines :
les nitrosamines génériques : petites molécules, non spécifiques (ex : NDBA, NDEA, NDIPA, NDMA, NEIPA et NMBA1)
les nitrosamines spécifiques des principes actifs ou de leurs impuretés = NDSRI (Nitrosamine Drug-Related Impurity). Celles-ci englobent :
les impuretés issues de la nitrosation d’un principe actif = N-nitroso impurity (i.e.NO-API) ;
les impuretés issues de la nitrosation d’une impureté d’un principe actif.
En raison de leur structure, environ 40 % des principes actifs pharmaceutiques et environ 30 % des impuretés de principes actifs pharmaceutiques seraient des précurseurs de nitrosamines2.
Leur détection dans plusieurs médicaments de la famille des « sartans » (valsartan, candésartan, irbésartan, losartan et olmésartan) a mis en lumière un risque jusque-là sous-estimé : la possibilité de formation de ces impuretés au cours de la synthèse, pendant la fabrication, ou même lors du stockage de la substance active et du produit fini.
2. Un cadre réglementaire renforcé
Face à l’ampleur du problème, les autorités – EMA, FDA, Santé Canada– ont rapidement exprimé leurs attentes. En Europe, l’EMA et le CMDh ont publié une série de lignes directrices et de questions-réponses (Q&A) imposant aux titulaires d’AMM une démarche structurée en trois étapes (reprise par les autorités nationales compétentes de la plupart des Etats membres de l’UE) :
Étape 1 – Évaluation du risque & déclaration aux autorités
Chaque substance active et chaque produit fini doit faire l’objet d’une analyse approfondie des sources potentielles de nitrosamines telles qu’identifiées dans le document Q&A (EMA/409815/2020).
Étape 2 – Analyses confirmatoires & déclaration aux autorités
En cas de risque identifié, des méthodes validées doivent être mises en œuvre pour confirmer ou infirmer la présence de nitrosamines.
Étape 3 – CAPA court, moyen, long terme
Si une nitrosamine est détectée à des teneurs supérieures à 10 % de l’AL (la limite acceptable) calculée sur la base de l’AI (la dose acceptable), l’industriel doit proposer des mesures correctives (CAPA), par exemple :
modification du procédé de fabrication,
changement de fournisseur de matière première,
changement du conditionnement primaire,
renforcement des contrôles,
mise à jour du dossier réglementaire.
Ces exigences s’appliquent aux médicaments chimiques, biologiques, à base de plantes et radiopharmaceutiques.
Le Q&A (EMA/409815/2020)décrit quand et comment soumettre les résultats des rapports des étapes 1 et 2. Les templates permettant de formaliser ces déclarations sont disponibles sur les sites de l’EMA, du CMDh et des autorités nationales compétentes européennes.
La conformité à ces exigences est l’objet de contrôles réguliers par les autorités, y compris lors des inspections.
3. Définition des paramètres et tests toxicologiques
Les définitions des paramètres et tests toxicologiques suivant sont détaillés dans l’ICH M7 “Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk” (Rev2) :
AI = Acceptable Intake= Dose Acceptable – en ng/jour
L’AI est la quantité maximale admissible (en masse), habituellement pour un jour, calculée pour un produit donné, à partir de données de toxicité (notamment la TD50).
L’AI est associée à un risque de cancer négligeable.
L’AI est basée sur le pouvoir carcinogène d’un produit (TD50).
L’AI est applicable à toutes les voies d’administration d’un produit.
TD50 est la dose donnant 50% d’incidences de tumeur chez l’animal (équivalent à un niveau de probabilité de risque de cancer de 1:2).
AL = Acceptable Limit = Limite Acceptable = critère d’acceptation – en ng/g (ppb)
L’AL est la concentration maximale acceptable d’une impureté dans une substance médicamenteuse ou un produit pharmaceutique, dérivée de la dose acceptable (= AI) et de la dose journalière du médicament (= MDD pour Maximum Daily Dose).
4. Textes de référence européens : un cadre réglementaire en constante évolution
La gestion du risque nitrosamine s’appuie aujourd’hui sur un ensemble solide de textes européens, régulièrement mis à jour pour intégrer les avancées scientifiques et les retours d’expérience des autorités. L’EMA et l’EDQM ont publié plusieurs documents structurants, dont :
le “Questions & Answers on Nitrosamine Contamination”(EMA), véritable socle réglementaire détaillant les attentes en matière d’évaluation du risque, de tests confirmatoires et de stratégies de réduction/minimisation du risque ;
les lignes directrices du CMDh, qui précisent les obligations applicables aux titulaires d’AMM, notamment les échéances de soumission des évaluations de risque et des variations.
Ces textes constituent un référentiel incontournable en Europe pour les industriels, qui doivent non seulement s’y conformer, mais aussi assurer une veille active afin d’anticiper les évolutions réglementaires. La dynamique actuelle montre clairement que les autorités européennes continueront d’affiner leurs exigences, renforçant la nécessité d’une approche proactive et documentée.
5. Une vigilance qui s’inscrit dans la durée
Les autorités réglementaires européennes (EMA/CMDh/EDQM) n’ont eu de cesse d’ajuster leurs recommandations afin de maîtriser les risques, sur la base des connaissances scientifiques acquises sur le sujet « en temps réel »,ce qui a fortement déstabilisé toute la filière du médicament (fabricants de PA, fabricants de PF/TAMM).
Les textes réglementaires européens de référence évoluent régulièrement et rapidement ; il convient que les TAMM suivent leurs évolutions « en temps réel » pour s’adapter. Des outils (ex : questionnaires) sont mis à la disposition des TAMM pour les accompagner dans la réalisation des analyses de risques requises par les autorités.
Les industriels doivent donc maintenir une surveillance continue, intégrer ce risque dans leur système qualité et suivre les évolutions réglementaires.
Conclusion
Le risque nitrosamines a profondément transformé la manière dont l’industrie pharmaceutique aborde la maîtrise des impuretés dans les médicaments. Au-delà de la conformité réglementaire, il s’agit d’un enjeu de confiance et de sécurité pour les patients. Les entreprises qui adoptent une approche proactive, scientifique et documentée seront les mieux armées pour répondre aux attentes des autorités et garantir la qualité de leurs produits.
Atessia accompagne les industriels dans leur stratégie de mise en conformité.
2. Schlingemann et al. J. Pharm Sci. 112 (2023), 1287 1304
Sources :
CMDh/412/2019: CMDh practical guidance for Marketing Authorisation Holders of nationally authorised products (incl. MRP/DCP) in relation to the Art. 5(3) Referral on Nitrosamines
EMA/409815/2020: « Questions and answers for marketing authorisation holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products.
EMA/144509/2025 : Nitrosamine impurities in human medicines
ICH M7(R2) Guideline on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk
La ligne directrice ICH Q12 « Guideline on technical and regulatory considerations for pharmaceutical product lifecycle management » vise principalement à fournir un cadre facilitant la gestion des modifications post‑approbation relatives au Module 3 du dossier d’Autorisation de mise sur le marché (AMM), ou modifications « CMC », de manière plus prévisible et plus efficace.
Cette ligne directrice et ses annexes ont été adoptées par le Comité des Médicaments à Usage Humain (Committee for Medicinal Products for Human Use – CHMP) de l’EMA en janvier 2020.
Ce texte tend à promouvoir l’innovation et l’amélioration continue, et à renforcer l’assurance qualité et la fiabilité de l’approvisionnement des produits, y compris la planification proactive des ajustements de la chaîne logistique dans un contexte de transparence entre l’industrie pharmaceutique et les autorités de santé.
Il s’efforce également de promouvoir, pour les régulateurs (évaluateurs et inspecteurs), une meilleure compréhension des systèmes de management de la qualité (SMQ) pharmaceutique des demandeurs pour la gestion des modifications CMC post-AMM en vue de minimiser les variations réglementaires dans un contexte international.
Enfin, il complète les lignes directrices ICH Q8 à ICH Q11 offrant des opportunités pour une approche basée sur la connaissance scientifique et le risque pour évaluer les changements tout au long du cycle de vie du médicament :
ICH Q8 « Pharmaceutical Development » / ICH Q11 « Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) » : se concentrent sur les premières phases du cycle de vie du produit (développement, enregistrement et lancement) ;
ICH Q9 « Quality Risk Management » (QRM) : décrit les principes et les outils pour la gestion du risque qualité pouvant être appliqués à différents niveaux ;
ICH Q10 « Pharmaceutical Quality System » (PQS) : décrit un modèle évolutif d’un système de gestion de la qualité pouvant être mis en œuvre tout au long du cycle de vie du produit.
ICH Q12 vise à démontrer comment une meilleure connaissance des produits et des procédés peut contribuer à déterminer plus précisément quelles modifications post‑approbation nécessitent effectivement une soumission réglementaire.
Pour ce faire, la version finale du texte adoptée par l’ICH en novembre 2019 présente 4 outils réglementaires et 4 facilitateurs réglementaires, accompagnés de principes directeurs, destinés à harmoniser au niveau mondial la gestion des modifications CMC post‑AMM (cf. Tableau 1).
Tableau 1 : Outils et des facilitateurs réglementaires ICH Q12
Outils réglementaires
Catégorisation des modifications CMC postérieures à l’autorisation
Facilitateurs réglementaires
Système Qualité Pharmaceutique (PQS) et Gestion de des Changements
Conditions Etablies (ECs)
Lien entre l’Evaluation Réglementaire et l’Inspection
Protocole de Gestion des Modifications Après Autorisation (PACMP)
Approches Structurées pour les Modifications CMC Après Autorisation Fréquentes
Document de Gestion du Cycle de vie du Produit (PLCM)
Approches en matière de Données de Stabilité pour Soutenir l’Evaluation des Modifications CMC
Vers une mise en œuvre facilitée des pleines possibilités offertes par ICH Q12
En mars 2020, soit deux mois après l’adoption de la ligne directrice ICH Q12 par le CHMP, la Commission Européenne et l’EMA ont publié une note explicative relative à la mise en œuvre avec des restrictions dans l’Union Européenne (i.e. l’ICH Q12 Step 5).
Au moment de la publication de cette note, des différences conceptuelles entre le contenu de l’ICH Q12 et le cadre juridique de l’UE ont été identifiées comme pouvant représenter un frein à la mise en œuvre totale de cette nouvelle ligne directrice en Europe ; ces différences ne permettant pas une application complète de la flexibilité opérationnelle et réglementaire définie dans l’ICH Q12.
En particulier, les approches scientifiques supplémentaires fondées sur le risque pour définir les conditions établies (Established Conditions – EC, cf. chapitre 3 de l’ICH Q12) et les catégories de notification associées, ainsi que le document de gestion du cycle de vie du produit (Product Lifecycle Management – PLCM, cf. chapitre 5 de l’ICH Q12), n’étaient pas considérées comme compatibles avec le cadre juridique l’UE relatif aux variations* en vigueur à cette époque.
Suite à la révision du règlement sur les modifications applicable depuis le 1er janvier 2025 en Europe, la Commission européenne a adopté et publié le 22 septembre 2025 la version finale des lignes directrices sur les détails des différentes catégories de modifications et le fonctionnement des procédures (applicables à partir du 15 janvier 2026). Ce règlement est ainsi désormais compatible avec l’ICH Q12 pour les sujets le concernant (cf. Tableau 2).
En ce qui concerne le système de qualité pharmaceutique (Pharmaceutical Quality System – PQS, cf. chapitre 6 de l’ICH Q12) et la gestion des changements, ainsi que le lien entre l’évaluation réglementaire et l’inspection (cf. chapitre 7 de l’ICH Q12), certaines clarifications supplémentaires concernant la démonstration et l’évaluation de l’efficacité du système de management de la qualité (SMQ) ainsi que la communication entre autorités réglementaires nécessitent encore une réflexion plus approfondie lors de la mise en œuvre de la ligne directrice (cf. Tableau 2).
Tableau 2 : Evolutions réglementaires européennes versus principaux outils/concepts ICH Q12
Principaux outils/concepts – ICHQ12
Ligne directrices variations (2013)
Ligne directrices variations (2025)
Commentaires
Chapitre 2 – Catégorisation des Modifications CMC Postérieures à l’Autorisation
Lignes directrices 2025 : ajout d’outils réglementaires additionnels compatibles ICH Q12 (QbD, PACMP, PLCM) dans cat. Q.I.e & Q.II.g
Chapitre 3 – Conditions Etablies (ECs)
Les ECs reflètent généralement les informations et caractéristiques de qualité soumises à variation.
Chapitre 4 – Protocole de Gestion des Modifications Après Autorisation (PACMP)
Lignes directrices 2025 : > Substance active : cat. Q.I.e.2 à Q.I.e.5 > Produit fini : cat. Q.II.g.2 à Q.II.g.5
Chapitre 5 – Document de Gestion du Cycle de vie du Produit (PLCM)
Lignes directrices 2025 : > Substance active : cat. Q.I.e.6 à Q.I.e.8 > Produit fini : cat. Q.II.g.6 à Q.II.g.8
Chapitre 6 – Système Qualité Pharmaceutique (PQS) et Gestion de des Changements
Chapitre 7 – Lien entre l’Evaluation Réglementaire et l’Inspection
–
–
clarifications attendues
Chapitre 8 – Approches Structurées pour les Modifications CMC Postérieures à l’Autorisation Fréquentes
–
–
clarifications attendues
Chapitre 9 – Approches en matière de Données de Stabilité pour Soutenir l’Evaluation des Modifications CMC
–
–
cf. draft ICH Q1 – clarifications attendues
Compatible // Pas compatible // – Pas de référence
Pour conclure, les évolutions de la réglementation européenne tendent à rendre effective la mise en œuvre de l’ICH Q12 afin de bénéficier de l’ensemble des progrès envisagés dans ce texte et ainsi faciliter la gestion des modifications CMC postérieures à l’autorisation de mise sur le marché de manière plus prévisible et plus efficace.
Par ailleurs, une future révision de la ligne directrice ICH Q12 sera à prévoir une fois la ligne directrice ICH M4Q(R2) « CTD on Quality » adoptée par le CHMP en Europe afin notamment d’en modifier l’annexe 1 (relative aux sections du CTD contenant des ECs).
* EC Variations Guidelines (2013) = Lignes directrices relatives aux caractéristiques des différentes catégories de modifications, au déroulement des procédures prévues aux chapitres II, II bis, III et IV du règlement (CE) n o 1234/2008 de la Commission du 24 novembre 2008 concernant l’examen des modifications des termes d’une autorisation de mise sur le marché de médicaments à usage humain et de médicaments vétérinaires et à la documentation à soumettre en vertu de ces procédures (2013/C 223/01)
** EC Variations Guidelines (2025)= Lignes directrices relatives aux caractéristiques des différentes catégories de modifications, au déroulement des procédures prévues aux chapitres II, II bis, III et IV du règlement (CE) no1234/2008 de la Commission concernant l’examen des modifications des termes d’une autorisation de mise sur le marché de médicaments à usage humain et à la documentation à soumettre en vertu de ces procédures (C/2025/5045)
La procédure centralisée est définie par le Règlement (CE) n° 726/2004 du Parlement européen et du Conseil du 31 mars 2004, établissant des procédures communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant l’Agence européenne des médicaments (EMA).
Cette procédure permet d’obtenir une autorisation de mise sur le marché (AMM) unique, délivrée par la Commission européenne, valable dans l’ensemble des États membres de l’Union européenne ainsi que dans les États membres de l’Espace Économique Européen (EEE) : l’Islande, le Liechtenstein et la Norvège.
1/ Champ d’application
La procédure centralisée est obligatoire pour certaines catégories de médicaments, à savoir :
les médicaments de thérapie innovante (advanced therapies)
les médicaments issus des biotechnologies
les médicaments contenant une nouvelle substance active non autorisée utilisé dans le traitement du syndrome d’immunodéficience acquise (SIDA), du cancer, des maladies neurodégénératives, du diabète, des maladies auto-immunes et autres dysfonctionnements immunitaires et des maladies virales
les médicaments désignés comme médicaments orphelins.
Elle est optionnelle pour d’autres catégories de médicaments qui, bien que ne relevant pas du champ obligatoire, peuvent présenter un intérêt à l’échelle européenne. Il s’agit :
des médicaments contenant une nouvelle substance active qui, au 20 mai 2004, n’était pas autorisée dans l’Union européenne
des médicaments constituant une innovation significative sur le plan thérapeutique, scientifique ou technique
des médicaments présentant un intérêt au niveau communautaire pour les patients.
Les médicaments génériques d’un médicament de référence autorisé dans l’Union peuvent également être éligibles à la procédure centralisée.
2/ Les acteurs impliqués
Le Comité des médicaments à usage humain (CHMP), organe scientifique de l’EMA, est au cœur de la procédure centralisée. Pour s’acquitter des tâches en matière de pharmacovigilance, le CHMP s’appuie sur l’évaluation scientifique et les recommandations du Comité pour l’évaluation des risques en matière de pharmacovigilance (PRAC).
Les principaux acteurs sont :
Le demandeur : le titulaire du dossier d’AMM.
Les rapporteurs et co-rapporteurs : deux États membres désignés par le CHMP pour évaluer le dossier et rédiger les rapports d’évaluation.
Le CHMP : responsable de l’examen scientifique et de la formulation d’un avis sur le rapport bénéfice/risque du médicament.
La Commission européenne : autorité compétente pour la décision finale d’AMM, sur la base de l’avis du CHMP.
3/ Calendrier
La durée totale d’évaluation scientifique est de 210 jours, hors arrêts d’horloge (clock-stops) permettant au demandeur de répondre aux questions du CHMP.
Pré-soumission Le demandeur peut organiser une réunion de pré-soumission (Pre-submission meeting) avec l’EMA. Ces réunions constituent une opportunité essentielle pour obtenir des avis procéduraux et réglementaires de l’Agence et s’assurer que le dossier est conforme aux exigences de la procédure centralisée.
Soumission et validation Le dossier complet est soumis via le portail de l’EMA. Une validation technique est réalisée, portant sur la structure du dossier (eCTD), ainsi qu’une validation du contenu administratif et réglementaire. Une fois ces validations terminées, l’évaluation scientifique commence officiellement le Jour 1 (J1).
1er tour d’évaluation (J1 à J120) Les rapporteurs et co-rapporteurs désignés par le CHMP réalisent leur première évaluation scientifique et rédigent un rapport d’évaluation préliminaire, consolidé avec les commentaires des autres membres du CHMP.
Une peer review est ensuite menée par le rapporteur et le co-rapporteur pour harmoniser les avis et finaliser la liste des questions à adresser au laboratoire. Le J120 marque le début de la période de clock-stop, pendant laquelle le laboratoire dispose généralement d’un délai pouvant aller jusqu’à trois mois pour préparer son document de réponses aux questions. Le temps de réponse n’est pas compté dans les 210 jours réglementaires.
2ème tour d’évaluation (J121 à J210)
Après la période de clock-stop, le CHMP reprend l’évaluation scientifique sur la base des réponses fournies par le laboratoire (J121). Cette phase permet de vérifier que toutes les questions ont été correctement traitées et de finaliser le rapport d’évaluation (joint assessment) (J150).
Si certaines questions restent en suspens ou si un point nécessite clarification, le CHMP peut organiser un second clock-stop, pendant lequel le laboratoire fournit les informations complémentaires (J180).
Après ce second clock-stop, une oral explanation (explication orale) peut être organisée pour donner suite à une requête du demandeur ou à la demande du CHMP (J181). Cette audience est généralement organisée lorsque le CHMP conserve des objections majeures et permet au laboratoire de répondre directement aux points critiques soulevés par le comité.
Une fois toutes les réponses aux questions reçues, le rapport peut être finalisé et le CHMP adopte son avis final sur la balance bénéfice/risque du médicament(J210). Cet avis peut être positif, adopté soit par consensus, soit à la majorité absolue des membres, ou négatif. En cas d’avis négatif, le laboratoire a la possibilité de faire appel (re-examination) de la décision conformément aux procédures de l’EMA.
Décision de la Commission européenne Lorsque l’avis du CHMP est positif, la Commission européenne dispose de 67 jours pour prendre la décision finale d’octroi de l’autorisation de mise sur le marché.
4/ En résumé
La procédure centralisée est la voie d’autorisation privilégiée et la plupart du temps obligatoire pour les médicaments innovants souhaitant une présence européenne. Elle garantit une évaluation scientifique harmonisée. Toutefois, sa complexité requiert une préparation stratégique solide et une expertise réglementaire confirmée.
ATESSIA accompagne les entreprises pharmaceutiques dans la définition de leur stratégie d’enregistrement, la préparation et la soumission des dossiers en procédure centralisée, ainsi que dans la gestion des interactions avec l’EMA et la Commission européenne.
Article rédigé par Lamya SAOUSSEN, Consultante Junior en Affaires Réglementaires et Communications Externes
Les médicaments dits « Over The Counter » (OTC) ne constituent pas, une catégorie juridique de médicaments en droit français ou européen. Cette notion, importée des pays anglo-saxons, désigne les médicaments accessibles directement aux patients dans les lieux de dispensation autorisés. Ils favorisent l’automédication responsable et l’autonomie du patient.
En France, depuis le 1er juillet 2008, certains médicaments de médications officinales (MMO) peuvent être rendus directement accessibles au public dans les officines, sous conditions strictes. Ils sont appelés de plusieurs façons : « médicaments d’automédication », « médicaments en libre accès » ou encore « médicaments en accès direct ».
Un espace dédié en officine
L’article R4235-55 du Code de la santé publique (CSP) autorise les pharmaciens d’officine à présenter les MMO dans un espace dédié, clairement identifié et situé à proximité immédiate des postes de dispensation et d’alimentation du dossier pharmaceutique.
Leur dispensation reste soumise au contrôle effectif du pharmacien, dans le respect de l’obligation de conseil qui lui est imposé par le Code de déontologie et les bonnes pratiques de dispensation.
Critères d’éligibilité pour une inscription sur la liste des MMO
Pour être présentés en accès direct au public, les médicaments doivent être inscrits sur une liste établie par le Directeur général de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM). Selon l’article R5121-202 du CSP, cette inscription est soumise aux conditions suivantes :
Le médicament ne doit pas être soumis à prescription médicale obligatoire (ce qui exclut les médicaments classés sur les listes I et II des substances vénéneuses, les stupéfiants, ainsi que les médicaments à prescription restreinte) ;
Les indications thérapeutiques, la durée de traitement et les informations de la notice doivent permettre une utilisation sécurisée par le patient, sur conseil du pharmacien et sans prescription médicale : les symptômes doivent être facilement reconnaissables, et ne doivent pas risquer de cacher une affection grave ;
Le conditionnement (poids, volume, nombre d’unités de prise) doit être adapté à la posologie et à la durée de traitement recommandées dans la notice ;
Le médicament ne doit pas faire l’objet d’une interdiction ou restriction de publicité auprès du public pour des raisons de santé publique ;
Il ne doit pas figurer sur les listes prévues par l’article L162-17 du Code de la sécurité sociale, ouvrant droit à un remboursement par l’assurance maladie.
C’est donc tout un travail d’adaptation de l’AMM qui est à concevoir pour changer le statut d’un médicament.
Une démarche volontaire pour les titulaires d’AMM
L’inscription d’un médicament sur la liste des MMO résulte d’une procédure contradictoire initiée sur démarche volontaire des titulaires d’autorisation de mise sur le marché (TAMM).
La demande est adressée par mail à la Direction réglementation et déontologie. Le dossier se compose d’un formulaire accessible sur le site de l’ANSM. Le délai d’instruction du dossier est approximativement de 30 jours.
Au préalable, les titulaires doivent s’assurer que l’AMM satisfait aux critères énoncées à l’article R5121-202 précité. À défaut, une modification préalable via une variation d’AMM de type II est requise.
Plusieurs scénarios sont possibles selon que :
Le médicament contient (ou non) une substance active ou une association déjà inscrite sur la liste des médicaments de médication officinale ;
L’indication du médicament est (ou non) prévue par la liste indicative desindications thérapeutiques acceptées pour un accès direct en officine, établies par l’ANSM.
A la suite de l’examen de la demande, et conformément à l’article R5121-203, le Directeur Général de l’ANSM peut, par décision motivée, refuser l’inscription d’un médicament sur la liste des MMO pour tout motif de santé publique et notamment en cas de réévaluation de la balance bénéfice risque de la spécialité concernée. Il peut également suspendre ou retirer un médicament de la liste si ce dernier ne satisfait plus aux critères de l’article R5121-202 ou pour tout motif lié à la santé publique.
Une publicité autorisée auprès du grand public
Compte tenu des critères imposés par l’article R5121-202, ces médicaments satisfont aux conditions imposées par l’article L5122-6 du CSP et peuvent donc faire l’objet de publicité auprès du Grand Public, sous réserve de l’obtention d’un visa GP.
Les recommandations ANSM précisent que la référence au fait qu’il s’agit d’un médicament de médication officinale reste possible sous réserve « qu’elle revêt un caractère sobre et informatif […] ». Cette mention ne peut constituer l’axe principal de la communication et ne doit pas être interprété dans sa forme comme « […] une accréditation ou un label officiels du message publicitaire ».
Les messages ou exergues ne doivent pas suggérer que le médicament est efficace ou sûr.
Conclusion : un équilibre entre accessibilité et sécurité
Le cadre réglementaire français permet aux pharmaciens d’officine de mettre à disposition directe du public certains médicaments non soumis à prescription médicale, à condition que leur AMM autorise une utilisation sécurisée sur conseil du pharmacien. Cette démarche, volontaire pour les titulaires d’AMM, s’inscrit dans un équilibre entre accessibilité des soins et sécurité des patients.
Atessia accompagne ses clients pour définir la stratégie et rédiger le dossier de passage en libre-accès.
Article rédigé par Arthur DI RUGGIERO, Consultant en Affaires réglementaires
La recherche médicale constitue un levier essentiel pour le développement de nouveaux traitements et pour l’amélioration continue de la prise en charge des patients.
En France, cette recherche est régie par des dispositions strictes visant à protéger les participants et à garantir l’intégrité de la personne humaine. La loi Jardé, adoptée en mars 2012 et pleinement appliquée depuis novembre 2016, constitue le cadre juridique pour les recherches impliquant les personnes humaines (RIPH).
En France, cette recherche est régie par des dispositions strictes visant à protéger les participants et à garantir l’intégrité de la personne humaine. La loi Jardé, adoptée en mars 2012 et appliquée depuis novembre 2016, est le cadre juridique pour les recherches impliquant les personnes humaines (RIPH).
Articulation entre la loi Jardé et le cadre européen des essais cliniques
Rappelons que les médicaments faisant l’objet d’essais cliniques sont encadrés en premier lieu par le règlement (UE) 536/2014 relatif aux essais cliniques (Clinical Trial Regulation – CTR), entré en vigueur le 31 janvier 2022. Ce règlement a abrogé et remplacé la directive 2001/20/CE. Depuis cette date, les essais cliniques sur les médicaments doivent être soumis via le Clinical Trials Information System (CTIS). Pour rappel, tout essai clinique disposant d’au moins un site investigateur encore actif en France au 31 janvier 2025 a dû faire l’objet d’une demande de transition vers le CTIS, par son promoteur. L’Avis aux promoteurs de l’ANSM apporte les aspects pratiques liés au dépôt de dossiers.
La loi Jardé ne se substitue pas au CTR, mais vient compléter le dispositif européen par des exigences nationales spécifiques, notamment en matière de protection des personnes, de consentement et d’information, de classification des recherches, et de procédures impliquant les Comités de Protection des Personnes (CPP).
D’autres dispositions réglementaires sont également à intégrer par les promoteurs, telles que les démarches sur la protection des données personnelles (RGPD, méthodologies de référence CNIL), les démarches relatives à l’utilisation de médicaments composés en tout ou partie d’organismes génétiquement modifiés (OGM) et les règles spécifiques applicables à certains produits de santé. En effet, selon la nature de l’objet de recherche, les dispositions applicables diffèrent. La loi Jardé couvre ainsi : les médicaments, les dispositifs médicaux (investigation clinique), les DMDIV (étude de performance), les produits de thérapie cellulaire, tissus, organes, les produits sanguins labiles (PSL), mais aussi certaines recherches portant sur les compléments alimentaires ou les cosmétiques.
Qu’est-ce que la loi Jardé ?
La loi Jardé, du nom du député Olivier Jardé, est un texte qui encadre les conditions dans lesquelles des recherches impliquant la personne humaine peuvent être menées en France. Elle remplace la loi Huriet-Sérusclat de 1988 et vise à renforcer la protection des participants tout en simplifiant et en harmonisant les procédures, en tenant compte du niveau de risque encouru.
Les principaux textes de référence incluent ainsi :
Les recherches organisées et pratiquées sur l’être humain en vue du développement des connaissances biologiques ou médicales sont désignées par les termes « recherche impliquant la personne humaine ».
Il existe trois types de RIPH :
Catégorie
Dispositions légales
Cadre
Catégorie 1 : Recherches interventionnelles comportant un risque pour les participants (intervention non justifiée par la prise en charge habituelle)
Articles L1121-1 à L1121-17 du CSP
Elles nécessitent une autorisation préalable de l’ANSM et un avis favorable d’un Comité de Protection des Personnes (CPP).
Catégorie 2 : Recherches interventionnelles avec des risques et des contraintes minimes *
Article L1121-1 à L1121-17 du CSP
Elles requièrent un avis favorable d’un CPP, mais pas d’autorisation de l’ANSM.
Elles requièrent un avis favorable d’un CPP, mais pas d’autorisation de l’ANSM.
* Les recherches portant sur des médicaments ne peuvent en principe pas relever de la catégorie 2, sauf cas très spécifiques définis réglementairement. Un arrêté donne les critères à respecter pour rester dans le champ des RIPH 2.
Quelles conséquences pour les industriels ?
Information et consentement des participants
L’objectif de la loi Jardé est de garantir la sécurité des participants. Une attention toute particulière est portée sur les notions de consentement libre et éclairé et d’information claire, compréhensible et loyale.
Les industriels, en tant que promoteurs, doivent s’assurer que les participants comprennent bien les enjeux, les procédures, les risques et contraintes et les bénéfices potentiels de l’étude. Ces exigences sont détaillées dans les articles L1122-1-1 à L1122-2 du CSP.
Détermination des autorités compétentes et procédures applicables
Les promoteurs doivent déterminer la catégorie de leur recherche dès la phase de conception, s’assurer qu’ils obtiennent les autorisations nécessaires (CPP et / ou ANSM) et définir la procédure applicable (CTR/CTIS pour les médicaments).
Interactions avec les CPP et l’ANSM
Les CPP sont les comités d’éthique français (Comités de Protection des Personnes) chargés d’évaluer la protection des participants, la qualité de l’information et du consentement et la balance bénéfice/risque. Les relations avec les CPP et l’ANSM sont essentielles pour la validation des projets de recherche. Une bonne communication et une soumission de dossiers complets sont nécessaires, conformément aux articles L1123-6 et L1123-7 du CSP. Un CPP en charge de l’évaluation du dossier est tiré au sort pour chaque RIPH. Le système d’information des recherches impliquant la personne humaine (SI RIPH 2G) permet de déposer un dossier de demande d’avis et obtenir la désignation aléatoire d’un CPP. La plateforme permet aussi de déclarer les volontaires sains participants à une étude.
Démarches et procédures
Préalablement au dépôt du dossier de demande d’autorisation (autorisation initiale et modification substantielle) et/ou d’avis de recherche impliquant la personne humaine, ou de recherche de soins courants, les promoteurs doivent obtenir un numéro d’enregistrement ID-RCB de la recherche. Ce numéro permet d’identifier chaque recherche réalisée en France. Pour une demande d’autorisation et d’avis de recherche interverventionnelle portant sur un médicament à usage humain, les promoteurs doivent obtenir à la place un numéro d’enregistrement de la recherche dans la base de données européenne CTIS (auparavant : EudraCT).
Ensuite, les promoteurs adresseront le dossier de demande d’autorisation de recherche biomédicale et/ou d’avis, par voie électronique, à l’ANSM et/ou au CPP, conformément aux arrêtés en vigueur fixant les formats de dossier propre à chaque type de recherche. Différents « Avis aux promoteurs » encadrent ces démarches selon la situation.
Conclusion
La loi Jardé assure ainsi la sécurité des participants aux recherches cliniques en France.
Pour les industriels de santé, comprendre et respecter ces règlementations est non seulement une obligation légale, mais aussi un gage de qualité des données générées en vue notamment de leur utilisation dans un dossier d’AMM.
En intégrant les exigences de la loi Jardé dans leurs processus, les industriels contribuent au développement de traitements innovants tout en garantissant des standards éthiques élevés, conformément aux exigences françaises et européennes sur ce sujet.
Atessia vous accompagne dans la mise en œuvre de ces process avec son expertise sur les essais cliniques.
Le règlement (UE) 2017/745 introduit de nouvelles exigences afin de renforcer la sécurité des patients et utilisateurs. Une des nouveautés de ce nouveau règlement est la création d’une base de données européenne dédiée aux informations sur les dispositifs médicaux appelée EUDAMED.
Cette plateforme sécurisée permettra :
d’accroître la transparence des informations sur les dispositifs médicaux avec un accès au grand public
une meilleure coordination entre les États membres dans la surveillance post commercialisation des dispositifs médicaux
EUDAMED est une plateforme sécurisée utilisée pour recueillir et partager des données relatives aux dispositifs médicaux mis sur le marché de l’Union européenne ainsi que ceux faisant l’objet d’investigation clinique.
Le règlement introduit de nouvelles exigences applicables aux différents acteurs pour EUDAMED.
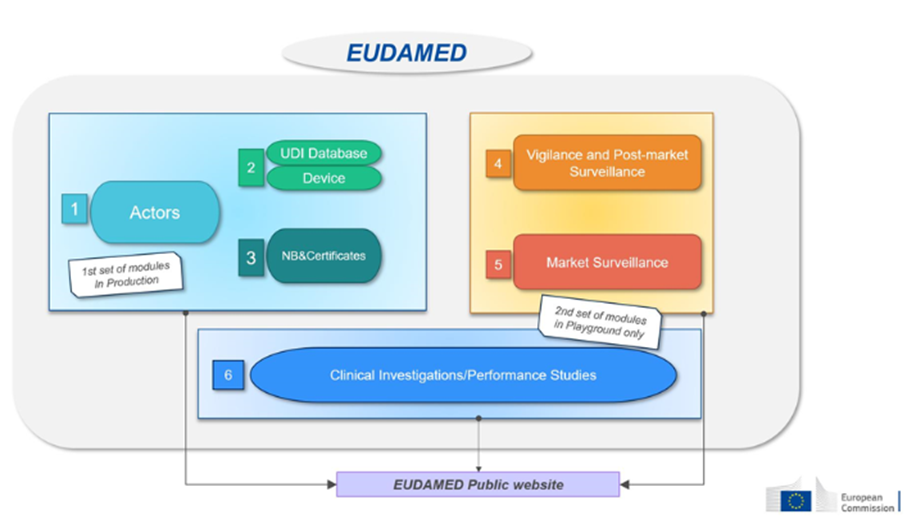
Cette base de données sera composée de 6 modules connectés les uns aux autres :
Les fabricants doivent soumettre dans EUDAMED le basic-IUD et les informations de tous les dispositifs qu’ils mettent sur le marché de l’UE.
Uniquement les fabricants
Enregistrement des dispositifs médicaux sous MDR
Aucune obligation pour les legacy devices (si enregistrement dans EUDAMED, il faudra faire un nouvel enregistrement pour les produits sous MDR, considérés comme de nouveaux produits)
Disponible sur base volontaire depuis octobre 2021 et est obligatoire à partir de T1 2026
Les organismes notifiés (ON) doivent enregistrer dans EUDAMED toute information concernant les certificats délivrés, suspendus, rétablis, retirés ou refusés et les autres restrictions imposées à ces certificats. Ces informations sont accessibles au public.
Organismes Notifiés
Disponible sur base volontaire depuis octobre 2021 et est obligatoire à partir de T1 2026
4-Vigilance
Module dédié à tous les rapports de vigilance et de surveillance post-commercialisation. -information de sécurité (Field Safety notice, FSN) -Actions correctives de sécurité (Field Safety Corrective Action, FSCA) -Rapport d’investigation des causes d’incident et mesures correctives (MIR) -Rapport de tendances (trend report) -Rapport périodique de sécurité (PSUR)
Fabricants
Sera obligatoire à partir de T4 2027
5-Surveillance du marché
La coordination des actions de surveillance de marché entre les différentes autorités compétentes.
Ce module concerne les enregistrements des investigations cliniques (DM) et études de performance (DMDIV). Rapport et résumé d’investigation clinique Evènement indésirable grave survenu pendant les investigations cliniques
Promoteurs
Sera obligatoire à partir de T4 2027
Source : Commission européenne
Et les distributeurs ?
Le MDR n’impose aucune exigence aux distributeurs concernant EUDAMED. Ils n’ont donc aucun accès sécurisé dans EUDAMED et ils ont uniquement l’accès grand public. Certains pays peuvent cependant définir des exigences supplémentaires, c’est le cas de la France qui demande aux distributeurs de s’enregistrer via le formulaire ANSM.
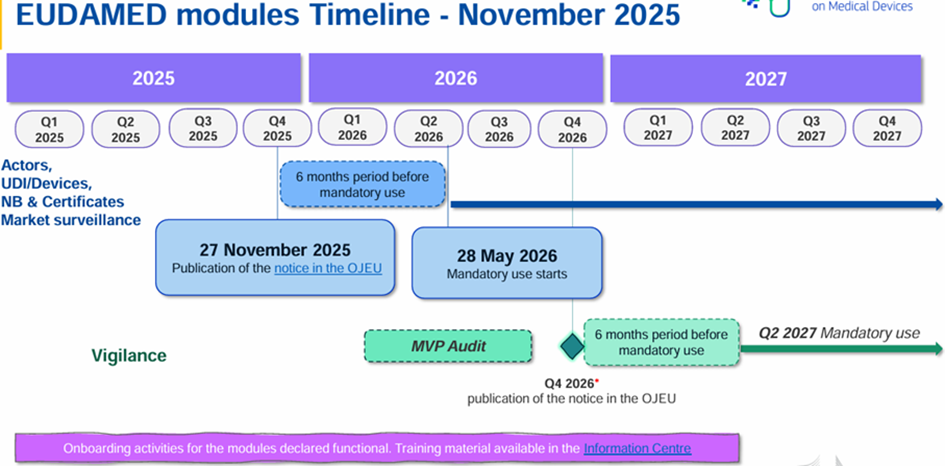
Calendrier de déploiement EUDAMED
En octobre 2019, la Commission européenne avait annoncé le report du lancement d’EUDAMED de 2 ans à mai 2022.
Certains modules sont déjà disponibles et peuvent être utilisés volontairement. Un projet de roadmap est sorti le 10 juillet 2024 indiquant un déploiement total d’EUDAMED prévu au deuxième trimestre 2027.
La Commission européenne a publié ce 27 novembre 2025 une décision (UE) 2025/2371 concernant la fonctionnalité et le respect des spécifications pour les modules EUDAMED :
Module « enregistrement des opérateurs économiques » (ACT module)
Module « enregistrement des dispositifs médicaux et base de données UDI » (UDI/DEV module)
Module « certificats et organismes notifiés » (NB/CRF module)
Module « surveillance sur le marché » (MSU module)
Ainsi ces 4 modules sont donc opérationnels et ils seront obligatoires à partir du 28 mai 2026 (soit 6 mois après la publication au JOUE) selon le règlement (UE) 2024/1860.
Pour les 2 modules restants, ils ne sont pour le moment pas disponibles :
Module investigations cliniques/studes de performance (CI/PS module)
Module surveillance post-marché et vigilance (VGL module)
Q&A“on practical aspects related to the implementation of the gradual roll-out of Eudamed pursuant to the MDR and IVDR, as amended by Regulation (EU) 2024/1860 amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards a gradual roll-out of Eudamed, the obligation to inform in case of interruption or discontinuation of supply, and transitional provisions for certain in vitro diagnostic medical devices” (22/11/2024)
USER GUIDES afin d’enregistrer les informations dans EUDAMED
Commission européenne – The EUDAMED four first modules will be mandatory to use as from 28 May 2026 (27/11/2025)
JOUE – Décision (UE) 2025/2371 de la Commission du 26 novembre 2025 relative à l’avis concernant la fonctionnalité et le respect des spécifications fonctionnelles de certains systèmes électroniques figurant dans la base de données européenne sur les dispositifs médicaux visée à l’article 34, paragraphe 1, du règlement (UE) 2017/745 du Parlement européen et du Conseil (27/11/2025)
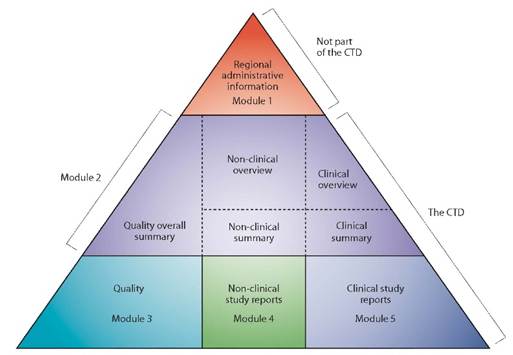
En Europe, les informations relatives à la qualité, à la sécurité et à l’efficacité du dossier d’Autorisation de Mise sur le Marché (AMM) des médicaments sont regroupées dans un format commun, appelé format CTD (Common Technical Document). Le format CTD s’applique dans toutes les régions reconnaissant les textes de l’ICH. Il est actuellement organisé selon cinq Modules : le Module 1 est spécifique à la région tandis que les Modules 2, 3, 4 et 5 sont communs à l’ensemble des régions (cf. figure 1).
Figure 1 : Le triangle du CTD
Le format CTD, décrit dans l’ICH M4, est devenu le format obligatoire pour les demandes d’AMM de nouveaux médicaments en Europe en juillet 2003. Avant sa mise en œuvre, les dossiers d’AMM répondaient aux exigences du format NtA Volume 2B (Edition 1998).
Pour les industriels, la mise en place du format CTD a permis d’éliminer la nécessité de reformater les informations à soumettre aux différentes autorités réglementaires de l’ICH.
Par la suite, la mise en place du format « eCTD » (electronic Common Technical Document), d’abord rendu obligatoire en Europe en 2007 pour les AMM en procédure centralisée, a révolutionné les pratiques réglementaires en harmonisant les soumissions électroniques auprès des autorités réglementaires de l’ICH, en remplaçant notamment les soumissions au format NeeS (Non-eCTD electronic Submission). Les informations relatives au format eCTD sont disponibles dans l’ICH M8.
Les raisons de la refonte de l’ICH M4Q(R1)
En Europe, le contenu du Module 2.3(QOS – Quality Overall Summary) et du Module 3 du dossier d’AMM répond aux exigences détaillées dans les lignes directrices ICH M4Q(R1) mises en œuvre en juillet 2003. Aucune révision de ce texte n’a été effectuée depuis plus de 20 ans, ce qui a conduit les législateurs à plusieurs constats :
Constat #1) La structure CTD traditionnelle n’est pas adaptée à la prise en charge des concepts modernes de qualité.
Depuis la publication de l’ICH M4Q(R1), de nouvelles lignes directrices ICH Q8 à ICH Q14 ont été élaborées, introduisant des concepts novateurs tels que la qualité par la conception (QbD – Quality by Design), la gestion des risques qualité (QRM – Quality Risk Management) et les approches fondées sur le cycle de vie (LCM – Life Cycle Management), ainsi que la fabrication continue (CM – Continous Manufacturing).
Le document ICH M4Q(R1) n’a pas été conçu pour tenir compte des nouveaux principes de qualité et leur intégration dans le format CTD actuel n’est pas aisée.
Constat #2) La structure CTD traditionnelle n’est pas adaptée à la prise en charge de l’évolution des technologies et des types de produits.
L’ICH M4Q(R1) a été conçu principalement pour les petites molécules conventionnelles et s’articule autour de la substance active (partie 32S) et du produit fini (partie 32P), avec des adaptations pour les produits biologiques. L’expérience a démontré que les produits complexes et les nouvelles modalités thérapeutiques (nanomédicaments, oligonucléotides et produits biologiques tels que les vaccins, les thérapies cellulaires et géniques et les produits issus de l’ingénierie tissulaire) ainsi que les produits combinés ne s’inscrivent souvent pas parfaitement dans ce cadre.
Constat #3) La structure CTD traditionnelle génère une ambiguïté dans l’organisation et le placement des informations.
Le format modulaire requis (i.e. résumé des données qualité versé dans le Module 2.3 et informations détaillées versées dans le Module 3) laisse place à des interprétations divergentes quant aux détails à inclure dans le Module 2.3 par rapport au Module 3 et conduit souvent à des répétitions d’informations. Une ambiguïté subsiste quant à l’emplacement des informations et aux références croisées entre les modules.
La gestion des mises à jour et des variations d’AMM tout le long du cycle de vie tout en conservant la cohérence dans la structure du CTD avec l’ICH M4Q(R1) n’est pas optimale.
Constat #4) La structure CTD traditionnelle laisse subsister des différences régionales.
Malgré l’harmonisation du format des dossiers au sein de l’ICH, des exigences supplémentaires spécifiques à certains pays/régions persistent souvent, réduisant ainsi l’avantage d’avoir un format unique.
Constat #5) Le format eCTD actuel ne permet pas d’intégrer les nouvelles exigences en matière de données électroniques et structurées.
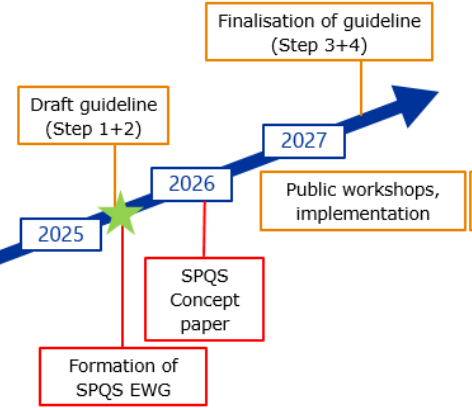
La tendance actuelle s’oriente vers des soumissions structurées, lisibles par des machines et vers l’utilisation de normes relatives aux données (ex : ISO IDMP 11615), en lien avec la future mise en œuvre du SPQS (Structured Product Quality Submission) par l’EMA qui conduira aux future lignes directrices ICH M16 (cf. figure 2).
Figure 2 : Interactions entre ICH M4Q(R2) et SPQS (future ICH M16)
L’ICH M4Q(R1) n’a pas été conçu pour de tels contenus structurés ce qui complique l’automatisation et empêche la réutilisation des données entre les soumissions.
Pour toutes ces raisons, l’ICH MQ4(R1) doit donc être repensée, pour permettre la gestion et la normalisation des données, et favoriser ainsi l’efficacité du processus d’examen et d’approbation des dossiers.
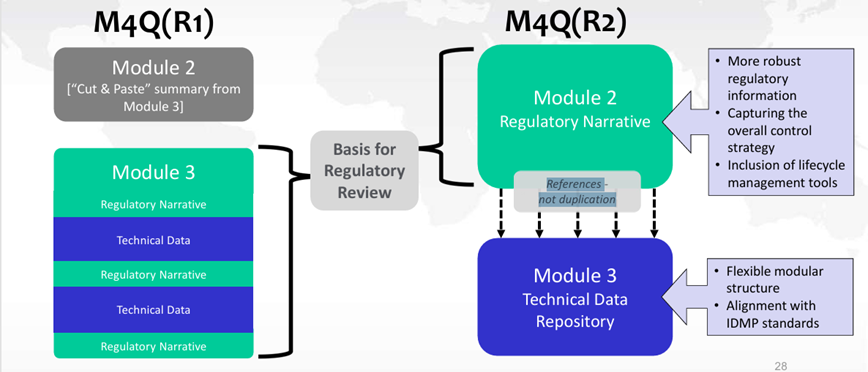
Aperçu de la révision d’ICH M4Q(R1) : cadre actuel versus cadre futur
Bien que des changements soient apportés à l’emplacement des informations dans les futurs Modules 2.3 et 3, ceux-ci ne modifient en rien les attentes réglementaires. Les données servant de base pour l’évaluation réglementaire, auparavant présentées dans le Module 3 figureront désormais dans le Module 2.3. Le Module 3 servira désormais de référentiel de données techniques (protocoles, rapports, données, …) (cf. figure 3).
Figure 3 : Changements de l’emplacement des données entre ICH M4Q(R1) et ICH M4Q(R2)
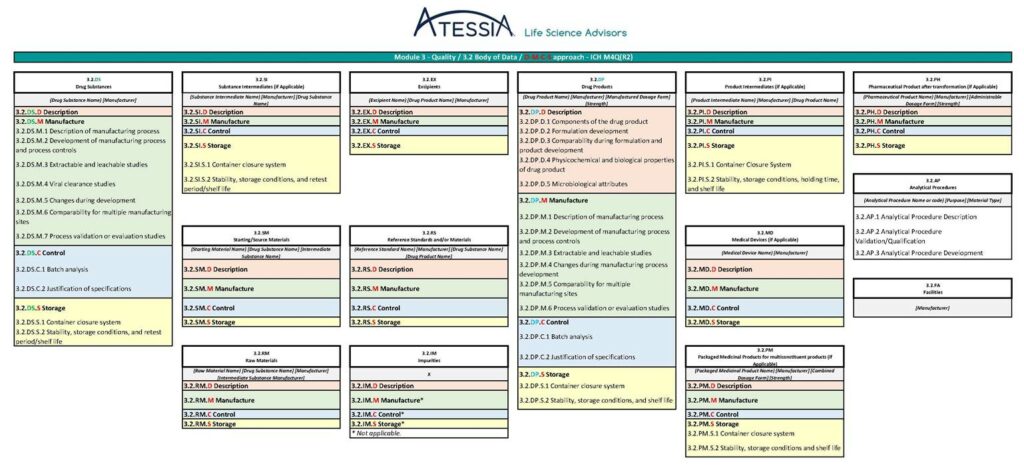
La future granularité du Module 2.3 et du Module 3, telle que pressentie dans la version « draft » du 14 mai 2025 de l’ICH M4Q(R2) est présentée en figure 4 et en figure 5.
Figure 4: Granularité du Module 2.3 selon ICH M4Q(R2) (1)
Figure 4 : Granularité du Module 2.3 selon ICH M4Q(R2) (2)
Figure 4 : Granularité du Module 2.3 selon ICH M4Q(R2) (3)
Figure 5 : Granularité du Module 3 selon ICH M4Q(R2)
Les informations essentielles sur la qualité (2.3.3 Core Quality Information (CQI)) :
– devront inclure toutes les informations assujetties à la gestion du cycle de vie conformément aux exigences régionales en matière de modifications post-AMM afin de garantir la qualité du produit.
– devront être conservées tout au long du cycle de vie du produit afin de garantir que les informations relatives à la qualité du produit restent à jour.
Les informations contenues dans les sections 2.3.1 (General Information), 2.3.2 (Overall Development and Overall Control Strategy), 2.3.4 (Development Summary and Justification), 2.3.5 (Product Lifecycle Management), 2.3.6 (Product Quality Benefit Risk) et le Module 3 seront des informations supportives, pouvant être modifiées ou complétées pour les soumissions post-AMM.
Dans la future ICH M4Q(R2), les informations seront regroupées dans des sous-sections spécifiques des matériaux/composants mis en œuvre dans la fabrication :
les substances médicamenteuses (DS),
les substances intermédiaires (SI),
les matières premières (RM),
les matières de départ (SM),
les excipients (EX),
les substances de référence (RS),
les impuretés (IM),
les médicaments (DP),
les produits intermédiaires (PI),
les médicaments conditionnés (PM),
les produits pharmaceutiques (PH) et
les dispositifs médicaux (MD).
Les informations relatives aux procédures analytiques et aux installations qui s’appliquent à tous les matériaux seront présentées dans des sections dédiées.
Chaque sous-section sera ensuite organisée selon la structure DMCS suivante :
Description / Description : identifie le matériau/composant et ses principales caractéristiques ;
Fabrication / Manufacture : décrit le procédé de production et les contrôles du procédé ;
Contrôle / Control : décrit les mesures de contrôle de la qualité telles que les spécifications ;
Stockage / Storage : fournit des informations sur le système de fermeture des contenants, la stabilité, les conditions de stockage et la période de recontrôle/durée de conservation.
Les relations entre le module 2.3 et le module 3 dans le contexte du modèle DMCS utilisé pour les matériaux/composants sont illustrées comme suit :
2.3.3 Core Quality Information
2.3.4 Development Summary and Justification
3.2 Body of Data
Informations relatives à la nature du produit et à ses principales caractéristiques, jugées nécessaires pour permettre l’autorisation de mise sur le marché et faciliter la gestion du cycle de vie
Résumé scientifique et fondé sur les risques et justifications relatives à la nature du matériau et à ses principales caractéristiques
Informations supportives, notamment des rapports et des données sur la nature du matériau et ses principales caractéristiques
Résumé de la caractérisation, développement de la formulation et justification
Données de caractérisation, développement de formulations et données justificatives
Fabrication
Description du processus de fabrication, IPC, paramètres critiques des procédés
Résumé du développement et de la validation/évaluation des procédés
Données relatives au développement et à la validation/évaluation des procédés
Contrôle
Spécifications
Aperçu de l’analyse par lots, justification des spécifications
Données d’analyse de lots et justification
Stockage
Description du conditionnement, des conditions de stockage et période de recontrôle/durée de conservation
Aperçu des études de stabilité, justification du conditionnement proposé
Choix du conditionnement et données relatives à la stabilité
Conclusion
En conclusion, l’ICH M4Q(R2) vise à favoriser l’harmonisation du contenu qualité des dossiers, afin de permettre, dans l’idéal, la soumission d’un dossier unique dans tous les pays membres de l’ICH.
Lorsque la loi l’exige, le demandeur devra fournir toute information supplémentaire spécifique à la région directement dans la section correspondante dans un document séparé, sous forme d’addendum au document de base harmonisé utilisé dans toutes les régions de l’ICH.
Atessia accompagne le secteur dans la rédaction des Modules 2.3 et 3.