

Transition RDM: Nouvelles Responsabilités et Changements Majeurs dans l’Univers des Dispositifs Médicaux
Depuis 1994, les dispositifs médicaux doivent répondre aux exigences des directives européennes 93/42/CEE et 90/385/CEE. La conformité à cette réglementation permet l’apposition du marquage CE et ainsi la libre circulation de dispositifs médicaux au sein de l’Union Européenne (UE).
L’Union Européenne (UE) a révisé cette réglementation par le règlement européen 2017/745/CE (RDM) qui est entré en vigueur le 26 mai 2021. L’adoption d’un règlement à la place d’une directive permet une uniformité du cadre réglementaire par son application directe.
Ce nouveau règlement donne de nouvelles responsabilités aux opérateurs économiques faisant partie de la chaine d’approvisionnement des dispositifs médicaux : fabricant, mandataire, importateur, distributeur.
Quels sont les principaux changements apportés par cette nouvelle réglementation (RDM) ?
- Re-désignation des organismes notifiés (ON) sous RDM ainsi qu’un contrôle des organismes notifiés (ON) par l’UE
- Une meilleure traçabilité grâce à l’IUD (Identifiant unique des dispositifs, UDI an anglais)
- L’inclusion de certains dispositifs esthétiques présentant les mêmes caractéristiques et le même profil de risque que des dispositifs médicaux relevant du champ d’application du règlement
- Une transparence améliorée grâce au système EUDAMED, la base de données sur les dispositifs médicaux
- Des exigences renforcées au niveau des données cliniques et sur la surveillance post commercialisation.
Quelles périodes de transition vont s’appliquer ?
Initialement, le RDM devait entrer en vigueur le 26 mai 2020. Dans le contexte de crise sanitaire du COVID de 2020, l’UE a décidé de modifier cette date via le règlement (UE) 2020/561.
La date d’application fut ainsi reportée au 26 mai 2021. A noter une période de transition pour les fabricants ayant des dispositifs médicaux déjà présents sur le marché européen « LEGACY DEVICES » jusqu’au 26 mai 2024 (fin de l’émission des certificats sous directive) et une mise à disposition de ces dispositifs médicaux sur le marché jusqu’au 26 mai 2025. En revanche, pour les dispositifs médicaux de classe I et les nouveaux dispositifs médicaux qui entrent sur le marché européen, ils doivent depuis 26 mai 2021 être conformes au RDM.
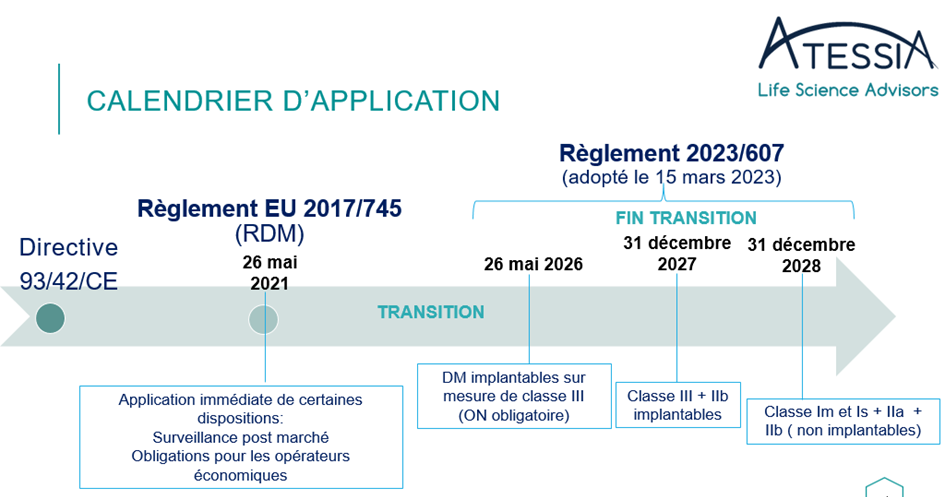
Le 20 mars 2023, l’UE avait évalué un risque de pénurie potentiel pour de nombreux DM et a ainsi adopté le règlement EU 2023/607 modifiant le règlement (UE) 2017/745 en ce qui concerne les dispositions transitoires (Article 120).
Les dates d’extension des certificats sous la directive 93/43/CE sont ainsi prolongées en fonction de la classe du dispositif médical :
Néanmoins les fabricants visés par cette extension doivent remplir des conditions :
1/ Les dispositifs continuent d’être conformes à la directive 90/385/CEE ou à la directive 93/42/CEE, selon le cas ;
2/ Il n’y a pas de modification significative de la conception et de la destination ;
3/ Les dispositifs ne présentent pas de risque inacceptable pour la santé ou la sécurité des patients, des utilisateurs ou d’autres personnes, ou compte tenu d’autres aspects liés à la protection de la santé publique ;
4/ Au plus tard le 26 mai 2024, le fabricant a mis en place un système de gestion de la qualité (art 10 (9)) ;
5/Le fabricant ou son mandataire a introduit auprès d’un organisme notifié une demande formelle d’évaluation de la conformité selon le RDM d’un dispositif médical ou d’un dispositif destiné à le remplacer au plus tard le 26 septembre 2024. L’organisme notifié et le fabricant ont signé un accord écrit.
Cas particulier pour les dispositifs de classe III implantables sur mesure – période transitoire introduite
L’évaluation de la conformité des dispositifs de classe III implantables sur mesure nécessite une évaluation par un ON.
Les DM de classe III implantables sur mesure peuvent être mis sur le marché sans le certificat correspondant jusqu’au 26 mai 2026, à condition que le fabricant ait déposé une demande auprès d’un ON désigné au titre du RDM avant le 26 mai 2024 et qu’il ait signé un contrat avec cet ON avant le 26 septembre 2024.
Les exigences du RDM applicables dès 26 mai 2021
Cependant, il est important de préciser que certaines exigences du RDM s’appliquent depuis le 26 mai 2021, comme les exigences relatives à la surveillance après commercialisation, à la surveillance du marché, à la vigilance, et à l’enregistrement des opérateurs économiques.
Suppression de la date de mise à disposition
De plus, le règlement EU 2023/607 vient supprimer totalement la date limite de mise à disposition, permettant ainsi aux « LEGACY DEVICES » d’être disponibles sans date limite (en respectant toujours la date limite d’utilisation du dispositif médical). Cette suppression est également ajoutée pour les dispositifs médicaux de diagnostic in vitro commercialisés avant le 26 mai 2022 dans le règlement (UE) 2017/746 (IVDR).
Les certificats CE sous les directives restent-il toujours valides pendant cette période d’extension ?
Les certificats qui ont expiré avant l’entrée en vigueur du règlement 2023/607 (20 mars 2023) doivent seulement être considérés valides si :
-Soit avant la date d’expiration du certificat, le fabricant et ON ont signé un accord d’évaluation de conformité avant la date d’expiration du certificat
– Ou si une autorité compétente a accordé une dérogation
Les ON ne peuvent plus émettre ni modifier les certificats CE selon les directives depuis le 26 mai 2021. De ce fait ils sont réputés étendus sauf s’ils ont été retirés.
Les fabricants peuvent émettre une auto-déclaration confirmant qu’ils sont conformes aux conditions de la prolongation en indiquant la date de fin de la période de transition (les dispositifs médicaux couverts par l’extension doivent être clairement indiqués). A la demande du fabricant, l’ON peut également émettre une lettre de confirmation.
Il est important de rappeler que, malgré ces reports, les fabricants doivent dès à présent mettre en place des actions vers ce nouveau règlement. Le délai de soumission aux organismes notifiés peut prendre de 6 à 18 mois selon l’ON et le dispositif médical.
Atessia accompagne ses clients dans toutes les étapes de la mise en conformité au MDR.









