Dans la vie d’un laboratoire pharmaceutique, les déménagements sont des évènements relativement rares. Quand ils se produisent, ce sont à de multiples challenges que sont confrontées les équipes : Naviguer dans les méandres de la réglementation applicable aux établissements pharmaceutiques pour trouver la bonne démarche à effectuer, multiplicité des démarches proposées sur le site de l’ANSM, déclarations obligatoires, imbroglios juridiques, nécessaire précision des informations à fournir, respect des délais. Atessia, fort de son expérience de dizaines de projets menés en 8 ans d’existence et sous la conduite de ses experts, saura être votre boussole pour vous orienter rapidement et éviter les écueils synonymes de perte de temps.
Qui ? Des interlocuteurs divers, en interne et en externe !
Première difficulté : S’orienter parmi les différents interlocuteurs nécessaires pour la réussite du projet.
Tout d’abord parmi les instances officielles, l’ANSM est la première et principale autorité concernée. En charge d’autoriser, d’inspecter et de surveiller l’activité des établissements, elle est clé pour obtenir le sésame : l’autorisation de déménager.
Mais ce n’est pas tout, le greffe sera sollicité, il faudra également maîtriser les subtilités des services en ligne de l’Union Européenne (ex : EMA account management, SPOR OMS…).
Selon les cas, votre carnet d’adresses sera mis à contribution : de nombreux interlocuteurs internes ou partenaires seront sollicités : information médicale, pharmacovigilance, supply chain, ceux-ci pouvant se trouver dans la filiale ou à la maison mère… sans compter les services IT et les services généraux ainsi que leurs prestataires (architectes, maîtrise d’ouvrage…) qui seront également clés dans le montage du dossier.
Quoi faire ? Des procédures en pagaille, en amont et en aval !
Outre le cœur du dossier (demande d’ouverture auprès de l’ANSM), il vous faudra tel un chef d’orchestre coordonner une pléiade de démarches administratives pour déclarer les fermetures, les ouvertures, les modifications administratives. La clé ? La méthode, l’expérience, et l’art du change control !
Maximiser ses chances de succès : La force de l’expérience Atessia
L’équipe Atessia, multidisciplinaire et menée par un chef de projet expert, saura vous guider pour identifier les points à risque, et mettre l’accent sur ceux qui permettront de répondre aux mieux aux attentes de l’autorité. Objectif zéro questions de l’ANSM pour des délais optimisés !
Et ce n’est pas terminé ! Des conséquences réglementaires qui nécessitent une parfaite coordination…
Adresse du titulaire d’AMM, Adresse de l’exploitant : Selon le portefeuille produit (nombre d’AMM, diversité des procédures), c’est toute une stratégie qui devra être mise au point, le cas échéant validée avec la maison mère, et, en fonction de l’importance du portefeuille, discutée avec l’ANSM pour une parfaite synchronisation. Les équipes d’Atessia peuvent ici aussi vous apporter toutes leurs connaissances dans la stratégie, et sa mise en œuvre, y compris dans la validation des articles de conditionnement et la stratégie de dépôt des variations d’AMM.
Quand les erreurs se payent cher
Les projets de déménagement s’étalent sur plusieurs mois, mais une simple erreur administrative peut induire un retard de plusieurs mois avec les conséquences financières associées. Pour assurer vos deadlines, Atessia vous fera profiter de son expérience et vous évitera les situations imprévues.
L’importance d’une gestion de projet efficace
Fort de son expérience, Atessia dédie un chef de projet expérimenté à votre projet. Vous n’avez plus qu’à vous relaxer et vous laisser guider !
La procédure centralisée est définie par le Règlement (CE) n° 726/2004 du Parlement européen et du Conseil du 31 mars 2004, établissant des procédures communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant l’Agence européenne des médicaments (EMA).
Cette procédure permet d’obtenir une autorisation de mise sur le marché (AMM) unique, délivrée par la Commission européenne, valable dans l’ensemble des États membres de l’Union européenne ainsi que dans les États membres de l’Espace Économique Européen (EEE) : l’Islande, le Liechtenstein et la Norvège.
1/ Champ d’application
La procédure centralisée est obligatoire pour certaines catégories de médicaments, à savoir :
les médicaments de thérapie innovante (advanced therapies)
les médicaments issus des biotechnologies
les médicaments contenant une nouvelle substance active non autorisée utilisé dans le traitement du syndrome d’immunodéficience acquise (SIDA), du cancer, des maladies neurodégénératives, du diabète, des maladies auto-immunes et autres dysfonctionnements immunitaires et des maladies virales
les médicaments désignés comme médicaments orphelins.
Elle est optionnelle pour d’autres catégories de médicaments qui, bien que ne relevant pas du champ obligatoire, peuvent présenter un intérêt à l’échelle européenne. Il s’agit :
des médicaments contenant une nouvelle substance active qui, au 20 mai 2004, n’était pas autorisée dans l’Union européenne
des médicaments constituant une innovation significative sur le plan thérapeutique, scientifique ou technique
des médicaments présentant un intérêt au niveau communautaire pour les patients.
Les médicaments génériques d’un médicament de référence autorisé dans l’Union peuvent également être éligibles à la procédure centralisée.
2/ Les acteurs impliqués
Le Comité des médicaments à usage humain (CHMP), organe scientifique de l’EMA, est au cœur de la procédure centralisée. Pour s’acquitter des tâches en matière de pharmacovigilance, le CHMP s’appuie sur l’évaluation scientifique et les recommandations du Comité pour l’évaluation des risques en matière de pharmacovigilance (PRAC).
Les principaux acteurs sont :
Le demandeur : le titulaire du dossier d’AMM.
Les rapporteurs et co-rapporteurs : deux États membres désignés par le CHMP pour évaluer le dossier et rédiger les rapports d’évaluation.
Le CHMP : responsable de l’examen scientifique et de la formulation d’un avis sur le rapport bénéfice/risque du médicament.
La Commission européenne : autorité compétente pour la décision finale d’AMM, sur la base de l’avis du CHMP.
3/ Calendrier
La durée totale d’évaluation scientifique est de 210 jours, hors arrêts d’horloge (clock-stops) permettant au demandeur de répondre aux questions du CHMP.
Pré-soumission Le demandeur peut organiser une réunion de pré-soumission (Pre-submission meeting) avec l’EMA. Ces réunions constituent une opportunité essentielle pour obtenir des avis procéduraux et réglementaires de l’Agence et s’assurer que le dossier est conforme aux exigences de la procédure centralisée.
Soumission et validation Le dossier complet est soumis via le portail de l’EMA. Une validation technique est réalisée, portant sur la structure du dossier (eCTD), ainsi qu’une validation du contenu administratif et réglementaire. Une fois ces validations terminées, l’évaluation scientifique commence officiellement le Jour 1 (J1).
1er tour d’évaluation (J1 à J120) Les rapporteurs et co-rapporteurs désignés par le CHMP réalisent leur première évaluation scientifique et rédigent un rapport d’évaluation préliminaire, consolidé avec les commentaires des autres membres du CHMP.
Une peer review est ensuite menée par le rapporteur et le co-rapporteur pour harmoniser les avis et finaliser la liste des questions à adresser au laboratoire. Le J120 marque le début de la période de clock-stop, pendant laquelle le laboratoire dispose généralement d’un délai pouvant aller jusqu’à trois mois pour préparer son document de réponses aux questions. Le temps de réponse n’est pas compté dans les 210 jours réglementaires.
2ème tour d’évaluation (J121 à J210)
Après la période de clock-stop, le CHMP reprend l’évaluation scientifique sur la base des réponses fournies par le laboratoire (J121). Cette phase permet de vérifier que toutes les questions ont été correctement traitées et de finaliser le rapport d’évaluation (joint assessment) (J150).
Si certaines questions restent en suspens ou si un point nécessite clarification, le CHMP peut organiser un second clock-stop, pendant lequel le laboratoire fournit les informations complémentaires (J180).
Après ce second clock-stop, une oral explanation (explication orale) peut être organisée pour donner suite à une requête du demandeur ou à la demande du CHMP (J181). Cette audience est généralement organisée lorsque le CHMP conserve des objections majeures et permet au laboratoire de répondre directement aux points critiques soulevés par le comité.
Une fois toutes les réponses aux questions reçues, le rapport peut être finalisé et le CHMP adopte son avis final sur la balance bénéfice/risque du médicament(J210). Cet avis peut être positif, adopté soit par consensus, soit à la majorité absolue des membres, ou négatif. En cas d’avis négatif, le laboratoire a la possibilité de faire appel (re-examination) de la décision conformément aux procédures de l’EMA.
Décision de la Commission européenne Lorsque l’avis du CHMP est positif, la Commission européenne dispose de 67 jours pour prendre la décision finale d’octroi de l’autorisation de mise sur le marché.
4/ En résumé
La procédure centralisée est la voie d’autorisation privilégiée et la plupart du temps obligatoire pour les médicaments innovants souhaitant une présence européenne. Elle garantit une évaluation scientifique harmonisée. Toutefois, sa complexité requiert une préparation stratégique solide et une expertise réglementaire confirmée.
ATESSIA accompagne les entreprises pharmaceutiques dans la définition de leur stratégie d’enregistrement, la préparation et la soumission des dossiers en procédure centralisée, ainsi que dans la gestion des interactions avec l’EMA et la Commission européenne.
Article rédigé par Lamya SAOUSSEN, Consultante Junior en Affaires Réglementaires et Communications Externes
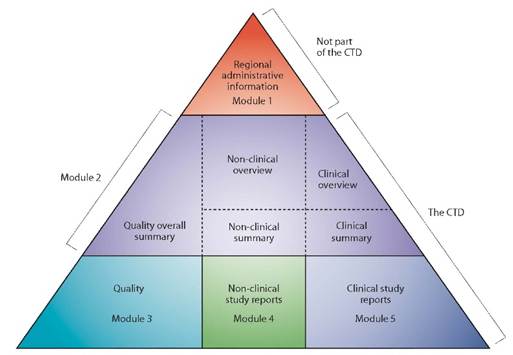
En Europe, les informations relatives à la qualité, à la sécurité et à l’efficacité du dossier d’Autorisation de Mise sur le Marché (AMM) des médicaments sont regroupées dans un format commun, appelé format CTD (Common Technical Document). Le format CTD s’applique dans toutes les régions reconnaissant les textes de l’ICH. Il est actuellement organisé selon cinq Modules : le Module 1 est spécifique à la région tandis que les Modules 2, 3, 4 et 5 sont communs à l’ensemble des régions (cf. figure 1).
Figure 1 : Le triangle du CTD
Le format CTD, décrit dans l’ICH M4, est devenu le format obligatoire pour les demandes d’AMM de nouveaux médicaments en Europe en juillet 2003. Avant sa mise en œuvre, les dossiers d’AMM répondaient aux exigences du format NtA Volume 2B (Edition 1998).
Pour les industriels, la mise en place du format CTD a permis d’éliminer la nécessité de reformater les informations à soumettre aux différentes autorités réglementaires de l’ICH.
Par la suite, la mise en place du format « eCTD » (electronic Common Technical Document), d’abord rendu obligatoire en Europe en 2007 pour les AMM en procédure centralisée, a révolutionné les pratiques réglementaires en harmonisant les soumissions électroniques auprès des autorités réglementaires de l’ICH, en remplaçant notamment les soumissions au format NeeS (Non-eCTD electronic Submission). Les informations relatives au format eCTD sont disponibles dans l’ICH M8.
Les raisons de la refonte de l’ICH M4Q(R1)
En Europe, le contenu du Module 2.3(QOS – Quality Overall Summary) et du Module 3 du dossier d’AMM répond aux exigences détaillées dans les lignes directrices ICH M4Q(R1) mises en œuvre en juillet 2003. Aucune révision de ce texte n’a été effectuée depuis plus de 20 ans, ce qui a conduit les législateurs à plusieurs constats :
Constat #1) La structure CTD traditionnelle n’est pas adaptée à la prise en charge des concepts modernes de qualité.
Depuis la publication de l’ICH M4Q(R1), de nouvelles lignes directrices ICH Q8 à ICH Q14 ont été élaborées, introduisant des concepts novateurs tels que la qualité par la conception (QbD – Quality by Design), la gestion des risques qualité (QRM – Quality Risk Management) et les approches fondées sur le cycle de vie (LCM – Life Cycle Management), ainsi que la fabrication continue (CM – Continous Manufacturing).
Le document ICH M4Q(R1) n’a pas été conçu pour tenir compte des nouveaux principes de qualité et leur intégration dans le format CTD actuel n’est pas aisée.
Constat #2) La structure CTD traditionnelle n’est pas adaptée à la prise en charge de l’évolution des technologies et des types de produits.
L’ICH M4Q(R1) a été conçu principalement pour les petites molécules conventionnelles et s’articule autour de la substance active (partie 32S) et du produit fini (partie 32P), avec des adaptations pour les produits biologiques. L’expérience a démontré que les produits complexes et les nouvelles modalités thérapeutiques (nanomédicaments, oligonucléotides et produits biologiques tels que les vaccins, les thérapies cellulaires et géniques et les produits issus de l’ingénierie tissulaire) ainsi que les produits combinés ne s’inscrivent souvent pas parfaitement dans ce cadre.
Constat #3) La structure CTD traditionnelle génère une ambiguïté dans l’organisation et le placement des informations.
Le format modulaire requis (i.e. résumé des données qualité versé dans le Module 2.3 et informations détaillées versées dans le Module 3) laisse place à des interprétations divergentes quant aux détails à inclure dans le Module 2.3 par rapport au Module 3 et conduit souvent à des répétitions d’informations. Une ambiguïté subsiste quant à l’emplacement des informations et aux références croisées entre les modules.
La gestion des mises à jour et des variations d’AMM tout le long du cycle de vie tout en conservant la cohérence dans la structure du CTD avec l’ICH M4Q(R1) n’est pas optimale.
Constat #4) La structure CTD traditionnelle laisse subsister des différences régionales.
Malgré l’harmonisation du format des dossiers au sein de l’ICH, des exigences supplémentaires spécifiques à certains pays/régions persistent souvent, réduisant ainsi l’avantage d’avoir un format unique.
Constat #5) Le format eCTD actuel ne permet pas d’intégrer les nouvelles exigences en matière de données électroniques et structurées.
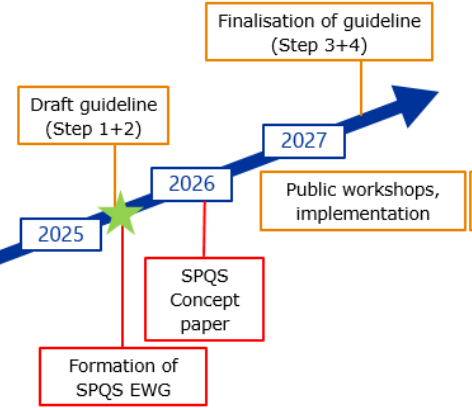
La tendance actuelle s’oriente vers des soumissions structurées, lisibles par des machines et vers l’utilisation de normes relatives aux données (ex : ISO IDMP 11615), en lien avec la future mise en œuvre du SPQS (Structured Product Quality Submission) par l’EMA qui conduira aux future lignes directrices ICH M16 (cf. figure 2).
Figure 2 : Interactions entre ICH M4Q(R2) et SPQS (future ICH M16)
L’ICH M4Q(R1) n’a pas été conçu pour de tels contenus structurés ce qui complique l’automatisation et empêche la réutilisation des données entre les soumissions.
Pour toutes ces raisons, l’ICH MQ4(R1) doit donc être repensée, pour permettre la gestion et la normalisation des données, et favoriser ainsi l’efficacité du processus d’examen et d’approbation des dossiers.
Aperçu de la révision d’ICH M4Q(R1) : cadre actuel versus cadre futur
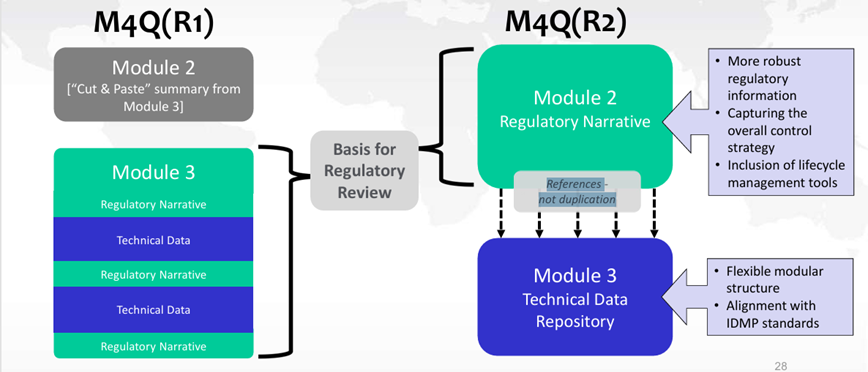
Bien que des changements soient apportés à l’emplacement des informations dans les futurs Modules 2.3 et 3, ceux-ci ne modifient en rien les attentes réglementaires. Les données servant de base pour l’évaluation réglementaire, auparavant présentées dans le Module 3 figureront désormais dans le Module 2.3. Le Module 3 servira désormais de référentiel de données techniques (protocoles, rapports, données, …) (cf. figure 3).
Figure 3 : Changements de l’emplacement des données entre ICH M4Q(R1) et ICH M4Q(R2)
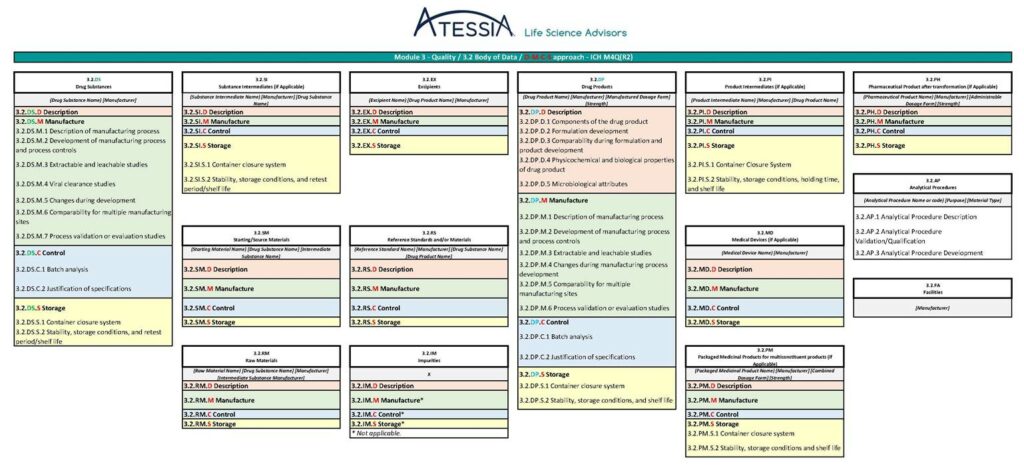
La future granularité du Module 2.3 et du Module 3, telle que pressentie dans la version « draft » du 14 mai 2025 de l’ICH M4Q(R2) est présentée en figure 4 et en figure 5.
Figure 4: Granularité du Module 2.3 selon ICH M4Q(R2) (1)
Figure 4 : Granularité du Module 2.3 selon ICH M4Q(R2) (2)
Figure 4 : Granularité du Module 2.3 selon ICH M4Q(R2) (3)
Figure 5 : Granularité du Module 3 selon ICH M4Q(R2)
Les informations essentielles sur la qualité (2.3.3 Core Quality Information (CQI)) :
– devront inclure toutes les informations assujetties à la gestion du cycle de vie conformément aux exigences régionales en matière de modifications post-AMM afin de garantir la qualité du produit.
– devront être conservées tout au long du cycle de vie du produit afin de garantir que les informations relatives à la qualité du produit restent à jour.
Les informations contenues dans les sections 2.3.1 (General Information), 2.3.2 (Overall Development and Overall Control Strategy), 2.3.4 (Development Summary and Justification), 2.3.5 (Product Lifecycle Management), 2.3.6 (Product Quality Benefit Risk) et le Module 3 seront des informations supportives, pouvant être modifiées ou complétées pour les soumissions post-AMM.
Dans la future ICH M4Q(R2), les informations seront regroupées dans des sous-sections spécifiques des matériaux/composants mis en œuvre dans la fabrication :
les substances médicamenteuses (DS),
les substances intermédiaires (SI),
les matières premières (RM),
les matières de départ (SM),
les excipients (EX),
les substances de référence (RS),
les impuretés (IM),
les médicaments (DP),
les produits intermédiaires (PI),
les médicaments conditionnés (PM),
les produits pharmaceutiques (PH) et
les dispositifs médicaux (MD).
Les informations relatives aux procédures analytiques et aux installations qui s’appliquent à tous les matériaux seront présentées dans des sections dédiées.
Chaque sous-section sera ensuite organisée selon la structure DMCS suivante :
Description / Description : identifie le matériau/composant et ses principales caractéristiques ;
Fabrication / Manufacture : décrit le procédé de production et les contrôles du procédé ;
Contrôle / Control : décrit les mesures de contrôle de la qualité telles que les spécifications ;
Stockage / Storage : fournit des informations sur le système de fermeture des contenants, la stabilité, les conditions de stockage et la période de recontrôle/durée de conservation.
Les relations entre le module 2.3 et le module 3 dans le contexte du modèle DMCS utilisé pour les matériaux/composants sont illustrées comme suit :
2.3.3 Core Quality Information
2.3.4 Development Summary and Justification
3.2 Body of Data
Informations relatives à la nature du produit et à ses principales caractéristiques, jugées nécessaires pour permettre l’autorisation de mise sur le marché et faciliter la gestion du cycle de vie
Résumé scientifique et fondé sur les risques et justifications relatives à la nature du matériau et à ses principales caractéristiques
Informations supportives, notamment des rapports et des données sur la nature du matériau et ses principales caractéristiques
Résumé de la caractérisation, développement de la formulation et justification
Données de caractérisation, développement de formulations et données justificatives
Fabrication
Description du processus de fabrication, IPC, paramètres critiques des procédés
Résumé du développement et de la validation/évaluation des procédés
Données relatives au développement et à la validation/évaluation des procédés
Contrôle
Spécifications
Aperçu de l’analyse par lots, justification des spécifications
Données d’analyse de lots et justification
Stockage
Description du conditionnement, des conditions de stockage et période de recontrôle/durée de conservation
Aperçu des études de stabilité, justification du conditionnement proposé
Choix du conditionnement et données relatives à la stabilité
Conclusion
En conclusion, l’ICH M4Q(R2) vise à favoriser l’harmonisation du contenu qualité des dossiers, afin de permettre, dans l’idéal, la soumission d’un dossier unique dans tous les pays membres de l’ICH.
Lorsque la loi l’exige, le demandeur devra fournir toute information supplémentaire spécifique à la région directement dans la section correspondante dans un document séparé, sous forme d’addendum au document de base harmonisé utilisé dans toutes les régions de l’ICH.
Atessia accompagne le secteur dans la rédaction des Modules 2.3 et 3.