Quelles sont les différentes options pour l’enregistrement d’une substance active dans un dossier d’Autorisation de Mise sur le Marché (AMM) en Europe ?
La description des données relatives à la substance active fait partie des éléments obligatoires devant figurer dans le Module 3 du dossier d’AMM. Ces éléments doivent être présentés selon le plan général décrit dans la Directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain.
Toutefois, si le fond des données attendues est précisé dans la réglementation européenne, la matérialisation de ces dernières dans le dossier d’AMM peut différer, selon l’option sélectionnée par le demandeur lors de l’agrément de son fabricant de substance active.
En Europe, l’enregistrement d’une substance inscrite par le demandeur d’AMM peut se faire via un ASMF/DPSA1 (cf. Directive 2001/83/EC, Annexe 1, Partie 1, Chapitre 3.2, Article (8)) ou via un CEP (cf. Directive 2001/83/EC, Annexe 1, Partie 1, Chapitre 3.2, Article (7)), le choix de la procédure étant laissé au fabricant de la substance active (aucune n’étant obligatoire).
Une autre possibilité est d’envisager la soumission, par le demandeur dans son dossier d’AMM, des données relatives à la substance active sous la forme d’une documentation scientifique dite « complète ».
On distingue différents cas de figures selon le type de substance active :
Type de substance active :
Voie documentaire applicable :
Nouvelle entité chimique (NtA, Volume 2A, Chapitre 1, Annexe I)
ASMF / DPSA ou documentation complète
Substance inscrite à la Pharmacopée européenne
ASMF / DPSA ou CEP ou documentation complète
Substance non-inscrite à la Pharmacopée européenne
ASMF / DPSA ou documentation complète
Dans cet article, nous nous attachons à expliciter les principales différences entre le CEP et l’ASMF pour enregistrer une substance active dans une autorisation de mise sur le marché en Europe.
Focus sur la procédure de CEP
Pour des informations complémentaires sur le CEP, consultez notre article de Blog « Qu’est-ce que les CEP ? ».
Focus sur la procédure d’ASMF / DPSA
Les requis pour la constitution d’un ASMF / DPSA en Europe sont décrits dans le guide de l’EMA intitulé « Final Guideline on Active Substance Master File Procedure (CHMP/QWP/227/02, EMEA/CVMP/134/02) » et le document de questions-réponses associé intitulé « Q&A on Active Substance master file (ASMF) » conjoint à l’EMA et au CMDh.
L’objectif principal de la procédure relative à l’ASMF est de permettre la protection de la propriété intellectuelle ou du « savoir-faire » confidentiel du fabricant de la substance active, tout en permettant au demandeur ou au titulaire de l’AMM d’assumer l’entière responsabilité du médicament, tout en garantissant la qualité et le contrôle de la qualité de la substance active.
Les autorités nationales compétentes / l’EMA ont ainsi accès à toutes les informations nécessaires pour évaluer l’adéquation de l’utilisation de la substance active dans le médicament.
Résumé des différences entre l’utilisation d’un CEP et d’un ASMF / DPSA
Un bref comparatif des avantages/inconvénients des procédures de CEP et d’ASMF / DPSA pour documenter la partie relative à la substance active dans un dossier d’AMM est présenté ci-après.
Critère
CEP
ASMF / DPSA
Confidentialité des données du fabricant de substance active
Très forte L’ensemble du Module 3.2.S est confidentiel (partagé avec l’EDQM uniquement, pas avec le TAMM)
Forte Module 3.2.S divisé en deux parties : > Partie fermée confidentielle > Partie ouverte non-confidentielle
Évaluation
Module 3.2.S évalué par EDQM (évaluation mutualisée) à Questions adressées uniquement au fabricant de la substance active
Parties ouverte et fermée évaluées par l’autorité où l’ASMF est déposé (évaluation non-mutualisée – sauf en cas de procédure de répartition des tâches « ASMF worksharing ») à Questions sur la partie fermée directement adressées au fabricant de la substance active ; questions sur la partie ouverte adressées au TAMM
Charge de travail pour le TAMM
Faible
Moyenne
Charge de travail pour le fabricant de substance active
Élevée au départ, faible ensuite
Moyenne
Gestion des variations d’AMM en lien avec la substance active
Très efficace : variations via des mises à jour du CEP
Moyenne
Quand est-ce recommandé ?
Substance avec monographie Ph. Eur. / commercialisation étendue de la substance active
Substance avec ou sans monographie Ph. Eur. / souhait de protection du savoir-faire
Les procédures réglementaires, les délais d’évaluation et les coûts associés sont différents.
Les consultants d’ATESSIA vous accompagnent dans la rédaction de vos dossiers de CEP, d’ASMF / DPSA et leur soumission auprès des autorités.
- EUR-Lex – Directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain
– EudraLex – Volume 2 – Notice to Applicants, Volume 2A, Chapter 1, Annex I
La ligne directrice ICH Q12 « Guideline on technical and regulatory considerations for pharmaceutical product lifecycle management » vise principalement à fournir un cadre facilitant la gestion des modifications post‑approbation relatives au Module 3 du dossier d’Autorisation de mise sur le marché (AMM), ou modifications « CMC », de manière plus prévisible et plus efficace.
Cette ligne directrice et ses annexes ont été adoptées par le Comité des Médicaments à Usage Humain (Committee for Medicinal Products for Human Use – CHMP) de l’EMA en janvier 2020.
Ce texte tend à promouvoir l’innovation et l’amélioration continue, et à renforcer l’assurance qualité et la fiabilité de l’approvisionnement des produits, y compris la planification proactive des ajustements de la chaîne logistique dans un contexte de transparence entre l’industrie pharmaceutique et les autorités de santé.
Il s’efforce également de promouvoir, pour les régulateurs (évaluateurs et inspecteurs), une meilleure compréhension des systèmes de management de la qualité (SMQ) pharmaceutique des demandeurs pour la gestion des modifications CMC post-AMM en vue de minimiser les variations réglementaires dans un contexte international.
Enfin, il complète les lignes directrices ICH Q8 à ICH Q11 offrant des opportunités pour une approche basée sur la connaissance scientifique et le risque pour évaluer les changements tout au long du cycle de vie du médicament :
ICH Q8 « Pharmaceutical Development » / ICH Q11 « Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) » : se concentrent sur les premières phases du cycle de vie du produit (développement, enregistrement et lancement) ;
ICH Q9 « Quality Risk Management » (QRM) : décrit les principes et les outils pour la gestion du risque qualité pouvant être appliqués à différents niveaux ;
ICH Q10 « Pharmaceutical Quality System » (PQS) : décrit un modèle évolutif d’un système de gestion de la qualité pouvant être mis en œuvre tout au long du cycle de vie du produit.
ICH Q12 vise à démontrer comment une meilleure connaissance des produits et des procédés peut contribuer à déterminer plus précisément quelles modifications post‑approbation nécessitent effectivement une soumission réglementaire.
Pour ce faire, la version finale du texte adoptée par l’ICH en novembre 2019 présente 4 outils réglementaires et 4 facilitateurs réglementaires, accompagnés de principes directeurs, destinés à harmoniser au niveau mondial la gestion des modifications CMC post‑AMM (cf. Tableau 1).
Tableau 1 : Outils et des facilitateurs réglementaires ICH Q12
Outils réglementaires
Catégorisation des modifications CMC postérieures à l’autorisation
Facilitateurs réglementaires
Système Qualité Pharmaceutique (PQS) et Gestion de des Changements
Conditions Etablies (ECs)
Lien entre l’Evaluation Réglementaire et l’Inspection
Protocole de Gestion des Modifications Après Autorisation (PACMP)
Approches Structurées pour les Modifications CMC Après Autorisation Fréquentes
Document de Gestion du Cycle de vie du Produit (PLCM)
Approches en matière de Données de Stabilité pour Soutenir l’Evaluation des Modifications CMC
Vers une mise en œuvre facilitée des pleines possibilités offertes par ICH Q12
En mars 2020, soit deux mois après l’adoption de la ligne directrice ICH Q12 par le CHMP, la Commission Européenne et l’EMA ont publié une note explicative relative à la mise en œuvre avec des restrictions dans l’Union Européenne (i.e. l’ICH Q12 Step 5).
Au moment de la publication de cette note, des différences conceptuelles entre le contenu de l’ICH Q12 et le cadre juridique de l’UE ont été identifiées comme pouvant représenter un frein à la mise en œuvre totale de cette nouvelle ligne directrice en Europe ; ces différences ne permettant pas une application complète de la flexibilité opérationnelle et réglementaire définie dans l’ICH Q12.
En particulier, les approches scientifiques supplémentaires fondées sur le risque pour définir les conditions établies (Established Conditions – EC, cf. chapitre 3 de l’ICH Q12) et les catégories de notification associées, ainsi que le document de gestion du cycle de vie du produit (Product Lifecycle Management – PLCM, cf. chapitre 5 de l’ICH Q12), n’étaient pas considérées comme compatibles avec le cadre juridique l’UE relatif aux variations* en vigueur à cette époque.
Suite à la révision du règlement sur les modifications applicable depuis le 1er janvier 2025 en Europe, la Commission européenne a adopté et publié le 22 septembre 2025 la version finale des lignes directrices sur les détails des différentes catégories de modifications et le fonctionnement des procédures (applicables à partir du 15 janvier 2026). Ce règlement est ainsi désormais compatible avec l’ICH Q12 pour les sujets le concernant (cf. Tableau 2).
En ce qui concerne le système de qualité pharmaceutique (Pharmaceutical Quality System – PQS, cf. chapitre 6 de l’ICH Q12) et la gestion des changements, ainsi que le lien entre l’évaluation réglementaire et l’inspection (cf. chapitre 7 de l’ICH Q12), certaines clarifications supplémentaires concernant la démonstration et l’évaluation de l’efficacité du système de management de la qualité (SMQ) ainsi que la communication entre autorités réglementaires nécessitent encore une réflexion plus approfondie lors de la mise en œuvre de la ligne directrice (cf. Tableau 2).
Tableau 2 : Evolutions réglementaires européennes versus principaux outils/concepts ICH Q12
Principaux outils/concepts – ICHQ12
Ligne directrices variations (2013)
Ligne directrices variations (2025)
Commentaires
Chapitre 2 – Catégorisation des Modifications CMC Postérieures à l’Autorisation
Lignes directrices 2025 : ajout d’outils réglementaires additionnels compatibles ICH Q12 (QbD, PACMP, PLCM) dans cat. Q.I.e & Q.II.g
Chapitre 3 – Conditions Etablies (ECs)
Les ECs reflètent généralement les informations et caractéristiques de qualité soumises à variation.
Chapitre 4 – Protocole de Gestion des Modifications Après Autorisation (PACMP)
Lignes directrices 2025 : > Substance active : cat. Q.I.e.2 à Q.I.e.5 > Produit fini : cat. Q.II.g.2 à Q.II.g.5
Chapitre 5 – Document de Gestion du Cycle de vie du Produit (PLCM)
Lignes directrices 2025 : > Substance active : cat. Q.I.e.6 à Q.I.e.8 > Produit fini : cat. Q.II.g.6 à Q.II.g.8
Chapitre 6 – Système Qualité Pharmaceutique (PQS) et Gestion de des Changements
Chapitre 7 – Lien entre l’Evaluation Réglementaire et l’Inspection
–
–
clarifications attendues
Chapitre 8 – Approches Structurées pour les Modifications CMC Postérieures à l’Autorisation Fréquentes
–
–
clarifications attendues
Chapitre 9 – Approches en matière de Données de Stabilité pour Soutenir l’Evaluation des Modifications CMC
–
–
cf. draft ICH Q1 – clarifications attendues
Compatible // Pas compatible // – Pas de référence
Pour conclure, les évolutions de la réglementation européenne tendent à rendre effective la mise en œuvre de l’ICH Q12 afin de bénéficier de l’ensemble des progrès envisagés dans ce texte et ainsi faciliter la gestion des modifications CMC postérieures à l’autorisation de mise sur le marché de manière plus prévisible et plus efficace.
Par ailleurs, une future révision de la ligne directrice ICH Q12 sera à prévoir une fois la ligne directrice ICH M4Q(R2) « CTD on Quality » adoptée par le CHMP en Europe afin notamment d’en modifier l’annexe 1 (relative aux sections du CTD contenant des ECs).
* EC Variations Guidelines (2013) = Lignes directrices relatives aux caractéristiques des différentes catégories de modifications, au déroulement des procédures prévues aux chapitres II, II bis, III et IV du règlement (CE) n o 1234/2008 de la Commission du 24 novembre 2008 concernant l’examen des modifications des termes d’une autorisation de mise sur le marché de médicaments à usage humain et de médicaments vétérinaires et à la documentation à soumettre en vertu de ces procédures (2013/C 223/01)
** EC Variations Guidelines (2025)= Lignes directrices relatives aux caractéristiques des différentes catégories de modifications, au déroulement des procédures prévues aux chapitres II, II bis, III et IV du règlement (CE) no1234/2008 de la Commission concernant l’examen des modifications des termes d’une autorisation de mise sur le marché de médicaments à usage humain et à la documentation à soumettre en vertu de ces procédures (C/2025/5045)
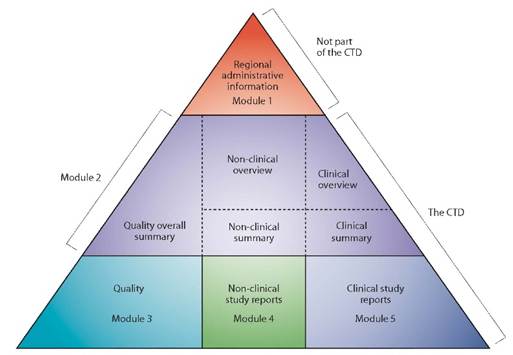
En Europe, les informations relatives à la qualité, à la sécurité et à l’efficacité du dossier d’Autorisation de Mise sur le Marché (AMM) des médicaments sont regroupées dans un format commun, appelé format CTD (Common Technical Document). Le format CTD s’applique dans toutes les régions reconnaissant les textes de l’ICH. Il est actuellement organisé selon cinq Modules : le Module 1 est spécifique à la région tandis que les Modules 2, 3, 4 et 5 sont communs à l’ensemble des régions (cf. figure 1).
Figure 1 : Le triangle du CTD
Le format CTD, décrit dans l’ICH M4, est devenu le format obligatoire pour les demandes d’AMM de nouveaux médicaments en Europe en juillet 2003. Avant sa mise en œuvre, les dossiers d’AMM répondaient aux exigences du format NtA Volume 2B (Edition 1998).
Pour les industriels, la mise en place du format CTD a permis d’éliminer la nécessité de reformater les informations à soumettre aux différentes autorités réglementaires de l’ICH.
Par la suite, la mise en place du format « eCTD » (electronic Common Technical Document), d’abord rendu obligatoire en Europe en 2007 pour les AMM en procédure centralisée, a révolutionné les pratiques réglementaires en harmonisant les soumissions électroniques auprès des autorités réglementaires de l’ICH, en remplaçant notamment les soumissions au format NeeS (Non-eCTD electronic Submission). Les informations relatives au format eCTD sont disponibles dans l’ICH M8.
Les raisons de la refonte de l’ICH M4Q(R1)
En Europe, le contenu du Module 2.3(QOS – Quality Overall Summary) et du Module 3 du dossier d’AMM répond aux exigences détaillées dans les lignes directrices ICH M4Q(R1) mises en œuvre en juillet 2003. Aucune révision de ce texte n’a été effectuée depuis plus de 20 ans, ce qui a conduit les législateurs à plusieurs constats :
Constat #1) La structure CTD traditionnelle n’est pas adaptée à la prise en charge des concepts modernes de qualité.
Depuis la publication de l’ICH M4Q(R1), de nouvelles lignes directrices ICH Q8 à ICH Q14 ont été élaborées, introduisant des concepts novateurs tels que la qualité par la conception (QbD – Quality by Design), la gestion des risques qualité (QRM – Quality Risk Management) et les approches fondées sur le cycle de vie (LCM – Life Cycle Management), ainsi que la fabrication continue (CM – Continous Manufacturing).
Le document ICH M4Q(R1) n’a pas été conçu pour tenir compte des nouveaux principes de qualité et leur intégration dans le format CTD actuel n’est pas aisée.
Constat #2) La structure CTD traditionnelle n’est pas adaptée à la prise en charge de l’évolution des technologies et des types de produits.
L’ICH M4Q(R1) a été conçu principalement pour les petites molécules conventionnelles et s’articule autour de la substance active (partie 32S) et du produit fini (partie 32P), avec des adaptations pour les produits biologiques. L’expérience a démontré que les produits complexes et les nouvelles modalités thérapeutiques (nanomédicaments, oligonucléotides et produits biologiques tels que les vaccins, les thérapies cellulaires et géniques et les produits issus de l’ingénierie tissulaire) ainsi que les produits combinés ne s’inscrivent souvent pas parfaitement dans ce cadre.
Constat #3) La structure CTD traditionnelle génère une ambiguïté dans l’organisation et le placement des informations.
Le format modulaire requis (i.e. résumé des données qualité versé dans le Module 2.3 et informations détaillées versées dans le Module 3) laisse place à des interprétations divergentes quant aux détails à inclure dans le Module 2.3 par rapport au Module 3 et conduit souvent à des répétitions d’informations. Une ambiguïté subsiste quant à l’emplacement des informations et aux références croisées entre les modules.
La gestion des mises à jour et des variations d’AMM tout le long du cycle de vie tout en conservant la cohérence dans la structure du CTD avec l’ICH M4Q(R1) n’est pas optimale.
Constat #4) La structure CTD traditionnelle laisse subsister des différences régionales.
Malgré l’harmonisation du format des dossiers au sein de l’ICH, des exigences supplémentaires spécifiques à certains pays/régions persistent souvent, réduisant ainsi l’avantage d’avoir un format unique.
Constat #5) Le format eCTD actuel ne permet pas d’intégrer les nouvelles exigences en matière de données électroniques et structurées.
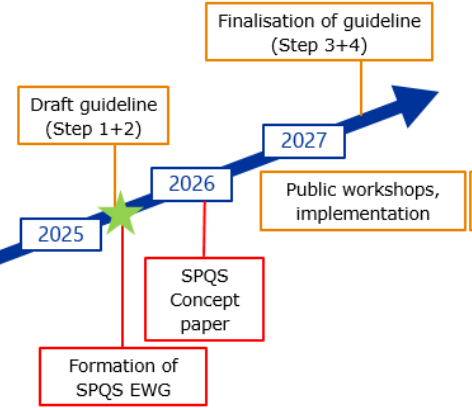
La tendance actuelle s’oriente vers des soumissions structurées, lisibles par des machines et vers l’utilisation de normes relatives aux données (ex : ISO IDMP 11615), en lien avec la future mise en œuvre du SPQS (Structured Product Quality Submission) par l’EMA qui conduira aux future lignes directrices ICH M16 (cf. figure 2).
Figure 2 : Interactions entre ICH M4Q(R2) et SPQS (future ICH M16)
L’ICH M4Q(R1) n’a pas été conçu pour de tels contenus structurés ce qui complique l’automatisation et empêche la réutilisation des données entre les soumissions.
Pour toutes ces raisons, l’ICH MQ4(R1) doit donc être repensée, pour permettre la gestion et la normalisation des données, et favoriser ainsi l’efficacité du processus d’examen et d’approbation des dossiers.
Aperçu de la révision d’ICH M4Q(R1) : cadre actuel versus cadre futur
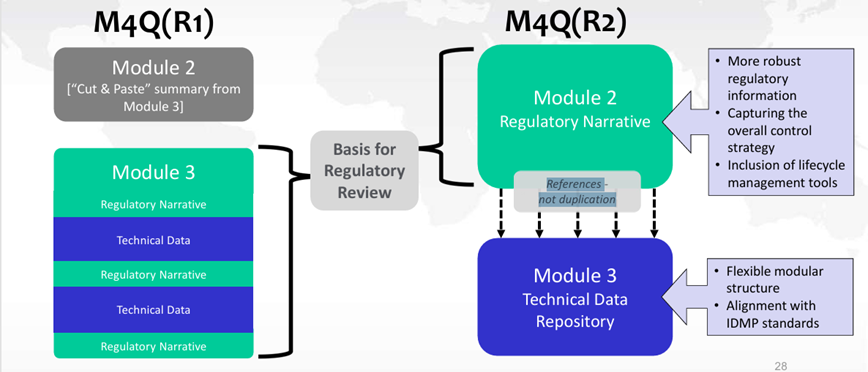
Bien que des changements soient apportés à l’emplacement des informations dans les futurs Modules 2.3 et 3, ceux-ci ne modifient en rien les attentes réglementaires. Les données servant de base pour l’évaluation réglementaire, auparavant présentées dans le Module 3 figureront désormais dans le Module 2.3. Le Module 3 servira désormais de référentiel de données techniques (protocoles, rapports, données, …) (cf. figure 3).
Figure 3 : Changements de l’emplacement des données entre ICH M4Q(R1) et ICH M4Q(R2)
La future granularité du Module 2.3 et du Module 3, telle que pressentie dans la version « draft » du 14 mai 2025 de l’ICH M4Q(R2) est présentée en figure 4 et en figure 5.
Figure 4: Granularité du Module 2.3 selon ICH M4Q(R2) (1)
Figure 4 : Granularité du Module 2.3 selon ICH M4Q(R2) (2)
Figure 4 : Granularité du Module 2.3 selon ICH M4Q(R2) (3)
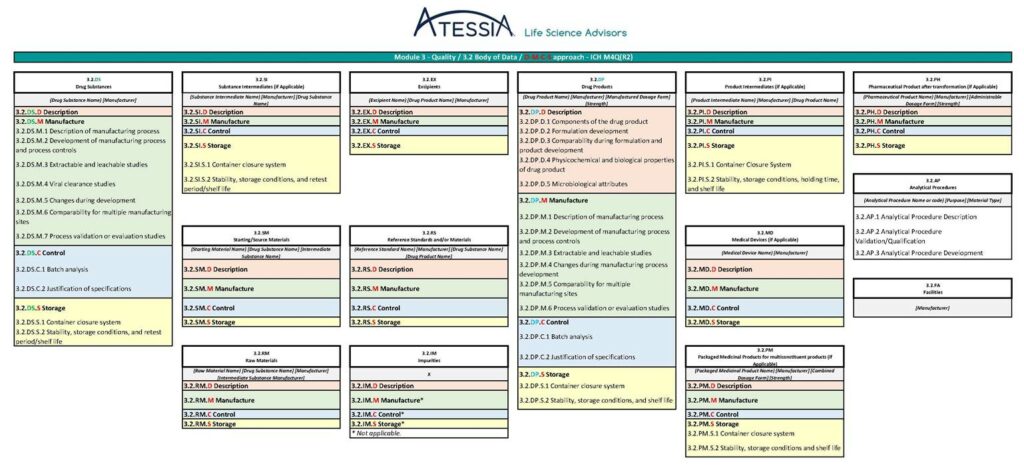
Figure 5 : Granularité du Module 3 selon ICH M4Q(R2)
Les informations essentielles sur la qualité (2.3.3 Core Quality Information (CQI)) :
– devront inclure toutes les informations assujetties à la gestion du cycle de vie conformément aux exigences régionales en matière de modifications post-AMM afin de garantir la qualité du produit.
– devront être conservées tout au long du cycle de vie du produit afin de garantir que les informations relatives à la qualité du produit restent à jour.
Les informations contenues dans les sections 2.3.1 (General Information), 2.3.2 (Overall Development and Overall Control Strategy), 2.3.4 (Development Summary and Justification), 2.3.5 (Product Lifecycle Management), 2.3.6 (Product Quality Benefit Risk) et le Module 3 seront des informations supportives, pouvant être modifiées ou complétées pour les soumissions post-AMM.
Dans la future ICH M4Q(R2), les informations seront regroupées dans des sous-sections spécifiques des matériaux/composants mis en œuvre dans la fabrication :
les substances médicamenteuses (DS),
les substances intermédiaires (SI),
les matières premières (RM),
les matières de départ (SM),
les excipients (EX),
les substances de référence (RS),
les impuretés (IM),
les médicaments (DP),
les produits intermédiaires (PI),
les médicaments conditionnés (PM),
les produits pharmaceutiques (PH) et
les dispositifs médicaux (MD).
Les informations relatives aux procédures analytiques et aux installations qui s’appliquent à tous les matériaux seront présentées dans des sections dédiées.
Chaque sous-section sera ensuite organisée selon la structure DMCS suivante :
Description / Description : identifie le matériau/composant et ses principales caractéristiques ;
Fabrication / Manufacture : décrit le procédé de production et les contrôles du procédé ;
Contrôle / Control : décrit les mesures de contrôle de la qualité telles que les spécifications ;
Stockage / Storage : fournit des informations sur le système de fermeture des contenants, la stabilité, les conditions de stockage et la période de recontrôle/durée de conservation.
Les relations entre le module 2.3 et le module 3 dans le contexte du modèle DMCS utilisé pour les matériaux/composants sont illustrées comme suit :
2.3.3 Core Quality Information
2.3.4 Development Summary and Justification
3.2 Body of Data
Informations relatives à la nature du produit et à ses principales caractéristiques, jugées nécessaires pour permettre l’autorisation de mise sur le marché et faciliter la gestion du cycle de vie
Résumé scientifique et fondé sur les risques et justifications relatives à la nature du matériau et à ses principales caractéristiques
Informations supportives, notamment des rapports et des données sur la nature du matériau et ses principales caractéristiques
Résumé de la caractérisation, développement de la formulation et justification
Données de caractérisation, développement de formulations et données justificatives
Fabrication
Description du processus de fabrication, IPC, paramètres critiques des procédés
Résumé du développement et de la validation/évaluation des procédés
Données relatives au développement et à la validation/évaluation des procédés
Contrôle
Spécifications
Aperçu de l’analyse par lots, justification des spécifications
Données d’analyse de lots et justification
Stockage
Description du conditionnement, des conditions de stockage et période de recontrôle/durée de conservation
Aperçu des études de stabilité, justification du conditionnement proposé
Choix du conditionnement et données relatives à la stabilité
Conclusion
En conclusion, l’ICH M4Q(R2) vise à favoriser l’harmonisation du contenu qualité des dossiers, afin de permettre, dans l’idéal, la soumission d’un dossier unique dans tous les pays membres de l’ICH.
Lorsque la loi l’exige, le demandeur devra fournir toute information supplémentaire spécifique à la région directement dans la section correspondante dans un document séparé, sous forme d’addendum au document de base harmonisé utilisé dans toutes les régions de l’ICH.
Atessia accompagne le secteur dans la rédaction des Modules 2.3 et 3.