Key points of the labelling of medicinal products in France
Introduction
Labelling of medicinal products in France is an important tool to ensure safe use by patients by providing easy to understand key user information. It is also a tool to fight against counterfeiting of medicines.
Development of labelling for medicines in France is precise task that must take into account multiple regulatory requirements.
1. Regulatory Framework
In addition to the information foreseen by annex IIIA of the Marketing authorisation, many other requirements arise from the French regulation.
-Identification
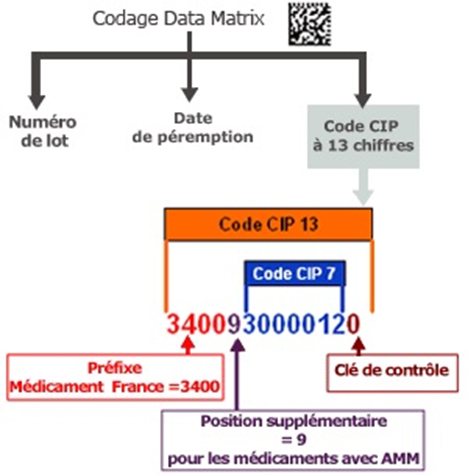
Packaging of all medicines must include the national administrative number called code CIP (code identifiant de presentation =
presentation identifier code) on both outer carton and inner packaging after the terms “Médicament autorisé n° …. » (13° of article R5121-138).
In addition, in line with the Directive 2001/83/CE and Delegated Regulation (EU) 2016/161, medicines must include an unique identifier (Articles R5121-138-1 and R5121-138-2) with exception to OTC products or POM products included in Annexe I of Delegated Regulation (EU) 2016/161 which are not requested to include the Unique identifier. In addition, packaging should bear an anti-tampering device on their packaging except for POM products included in Annexe I of Delegated Regulation (EU) 2016/161 .
In France, the Product Code (PC) (14-digit code) includes the national number (NN). The NN in France is the CIP and the PC corresponds to “0+code CIP” which should appear near the datamatrix and separately from the above mentioned “Médicament autorisé n° …. ».
-Legal Status
The legal status must be made available on both outer and inner packaging (17° of article R5121-138).
All medicines available on prescription are listed on List I or List II which determine how they can be delivered.
This classification must appear on the packaging with details as follow (Article R5132-15):
- an empty frame with a red or a green border depending on the List;
- In the coloured border, the text “respecter les doses prescrites*” in black font must be included;
- the below information must then be mentioned :
- “Liste I” or “Liste II”,
- “Uniquement sur ordonnance”**,
- “Ne pas avaler” (in case the product is not for oral, sublingual, perlingual or injectable administration)
In addition, if applicable, other information must appear in case:
- the medicine is classified as narcotic or psychotic,
- the medicine is subject to restricted or special prescription.
When medicines are contained in outer packaging that complies with the aforementioned provisions:
*The statement “Follow the prescribed doses” is not mandatory for ampoules or other small primary packaging where affixing this statement would not ensure optimal legibility of the information.
**The statement “Prescription only” is not mandatory for primary packaging containing only a single dose.
This information is made available in the prescription and delivery information approved by ANSM:
- within the MA for product authorised via NP, MRP or DCP,
- or within the blue box for product authorised via the CP.
-Pregnancy Pictogram
Requirement to include the Pregnancy pictogram has been introduced in 2017 and is included in Article R5121-139 of the French Public Health code. This pictogram placed on the outer packaging aims at providing patients with information on the risk of using the medicines during pregnancy or in case of childbearing potential. It concerns medicines with teratogenic or foetotoxic effects mentioned in the SmPC (sections 4.6 and 5.3).
Three situations are possible and the population in scope of the warning must be precisely mentioned below or on the right side of the pictogram:
| DANGER | PROHIBITED | VALPROATE |
 |  |  |
| The triangle must be equilateral and with a side of at least 1 cm. The circle must have a diameter of at least 1 cm. The colour of the form is red with white inner. The pregnant woman must be black. | ||
| Population in scope to be determined : « – l’adolescente ou la femme en âge de procréer, et sans contraception efficace ; – la femme enceinte ; – la femme enceinte à compter du [X]e mois de grossesse (lorsque la contre-indication porte sur une période précise de la grossesse). » | Population determined by law. | |
It is the responsibility of the MAH to determine the level of the pregnancy pictogram and the concerned population. It is highly recommended to ensure traceability of this pictogram determination and to ensure adequate stakeholders are involved.
These information will be then submitted to the French Agency upon declaration of marketing of the medicine.
-Pictogram regarding the effect on the ability to drive or use machines
Medicines which may reduce the ability to drive or operate machines must have a pictogram (warning triangle) (Article R5121-139). Its size is adapted to fit the label.
Since the ministerial order in 2008, 3 categories of pictograms have been identified for specific active substances (listed in ministerial decrees dated August 2008 and March 2017) in relation with the effect on the ability to drive. In addition, in case the active substance is not listed in the ministerial decree but is known to have such effect (based on section 4.7 of the SmPC), the MAH must include the neutral pictogram.
| Active substance listed in Ministerial Decree | Active substance not listed in Ministerial Decree but known to have effects on the ability to drive and use machines | ||
| Level 1 | Level 2 | Level 3 | |
 |  |  | |
-Pictogram for medicinal products containing ketoprofen as topical gel to avoid sun exposure of treated skin areas

-Other pictograms
Article R5121-139 also states that the outer packaging may include, in addition to the company’s distinctive mark, signs or pictograms explaining some of the labelling information as well as other information consistent with the SmPC if they are useful to patients and not promotional in nature.
-Logos foreseen in the environmental code
In addition to the pictograms foreseen in the French pharmaceutical legislation, it is necessary to consider the inclusion of the pictogram “INFOTRI” aiming at explain how to eliminate different elements of the medicinal product.
This INFOTRI logo is mandatory on all products intended for household use.
This logo is to be included on one of the packaging elements of medicinal products used by patients (outer packaging or PIL (except of centralised procedure)). This is not requested for medicinal products used by Healthcare Professionals.
2. Other texts to consider:
In addition to the regulatory texts, various recommendations from the French HAs and from the EU regulators must be taken into account to develop, review and approve artworks :
- French texts:
- Recommendation regarding the labelling of oral solid forms (outside homeopathic medicines),
- Core labelling for paracetamol containing medicines,
- EU texts:
- Notice To Applicants : Guideline on the packaging information of medicinal products for human use authorised by the union,
- CMDh “Blue – Box” requirements
3. Responsibilities
Validation of artwork for France is under the responsibility of the Chief Pharmaceutical Officer (CPO) of the Exploitant. The validation can be supported by the below proposed table.
| Registration procedure | What references to use | |
| National | Annex IIIB; French labelling recommandations List provided in Prescription and delivery part of the MA | Article R5121-139 for effect on the ability to drive or use machines + ministerial decree + content of section 4.7 of the SmPC Internal assessment for the determination of the pregnancy pictogram level + sections 4.6 and 5.3 of the SmPC; CIP code provided by ANSM Article of article R5121-138 (point 17°) Article R5132-15 INFOTRI |
| MR and DC procedures | Annex IIIB; French labelling recommandations Mock-up approved during the MR or DC procedure List provided in Prescription and delivery part of the MA | |
| Centralised | Annex IIIB; Blue box approved by the ANSM; Mock-up approved by the EMA QRD group | |
Atessia can support you for the development and review of artworks of medicinal products for the French Market.
Link : https://www.atessia.fr/en/our-services/registration-drafting-and-submitting/
Article written by Agathe DAUBISSE, Senior Regulatory Affairs Consultant