Understanding EUDAMED: The European Database on Medical Devices
The new regulation (EU) 2017/745 introduces new requirements to enhance the safety of patients and users. One of the novelties of this new regulation is the creation of a European database dedicated to information on medical devices called EUDAMED.
This database will allow:
- Increased transparency of information on medical devices with public access.
- Better coordination between Member States in the post-market surveillance of medical devices.
EUDAMED is a secure platform used to collect and share data related to medical devices placed on the European Union market, as well as those undergoing clinical investigation.
The regulation introduces new requirements for the various actors involved in EUDAMED.
This database will consist of six interconnected modules:
| Module : | Who needs to record information? | Accessible to the public | |
| 1-Actors | Economic operators must register as actors in EUDAMED and provide the required information. | – EU and third-country manufacturers, – Authorized representatives, – System/procedure pack producers, – Importers. | Available on a voluntary basis since December 2020 and will be mandatory from Q1 2026. |
| 2-Devices | Manufacturers must submit the basic-UDI and information of all devices they place on the EU market into EUDAMED. | Only manufacturers. Registration of medical devices under MDR. No obligation for legacy devices (if registered in EUDAMED, a new registration will be required for products under MDR, considered as new products). | Available on a voluntary basis since October 2021 and will be mandatory from Q1 2026. |
| 3- Notified Bodies (NB) and Certificates | Notified bodies must register in EUDAMED all information regarding issued, suspended, reinstated, withdrawn, or refused certificates and other restrictions imposed on these certificates. This information is accessible to the public. | Notified Bodies. | Available on a voluntary basis since October 2021 and will be mandatory from Q1 2026. |
| 4-Vigilance | Module dedicated to all vigilance and post-market surveillance reports. – Safety information (Field Safety Notice, FSN), – Field Safety Corrective Action (FSCA), – Investigation report of incident causes and corrective measures (MIR), – Trend report, – Periodic Safety Update Report (PSUR). | Manufacturer. | Will be mandatory from Q3 2026. |
| 5- Market Surveillance | Coordination of market surveillance actions between the various competent authorities. | Competent authority only. | Will be mandatory from Q1 2026. |
| 6- Clinical Investigation/Performance Studies (CI/PS): | This module concerns the registration of clinical investigations (MD) and performance studies (IVD). – Clinical investigation report and summary, – Serious adverse event during clinical investigations. | Sponsor. | Not yet available. |
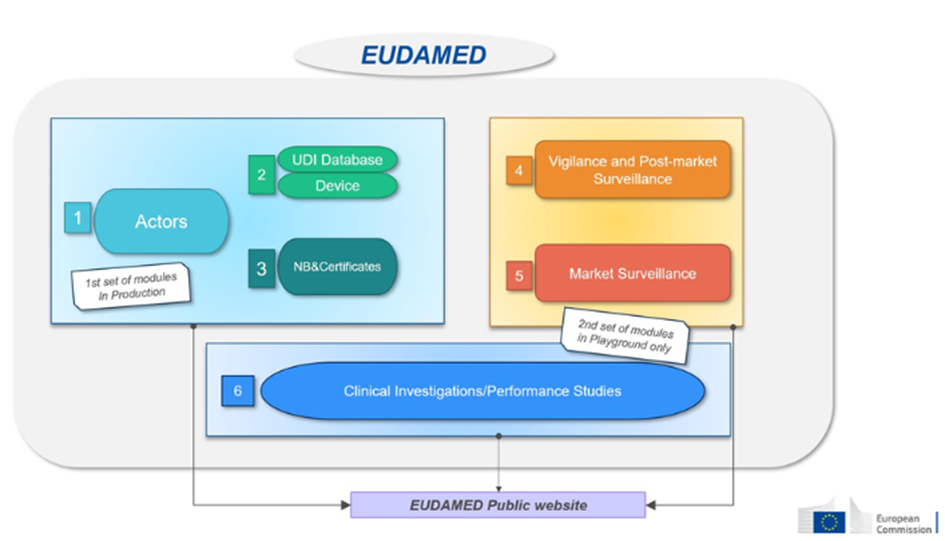
Source : European commission
And the Distributors?
The MDR imposes no requirements on distributors regarding EUDAMED. They have no secure access to EUDAMED and only have public access. However, some countries may set additional requirements, such as France, which requires distributors to register via the ANSM form.
EUDAMED Deployment Schedule
In October 2019, the European Commission announced a two-year postponement of EUDAMED’s launch to May 2022.
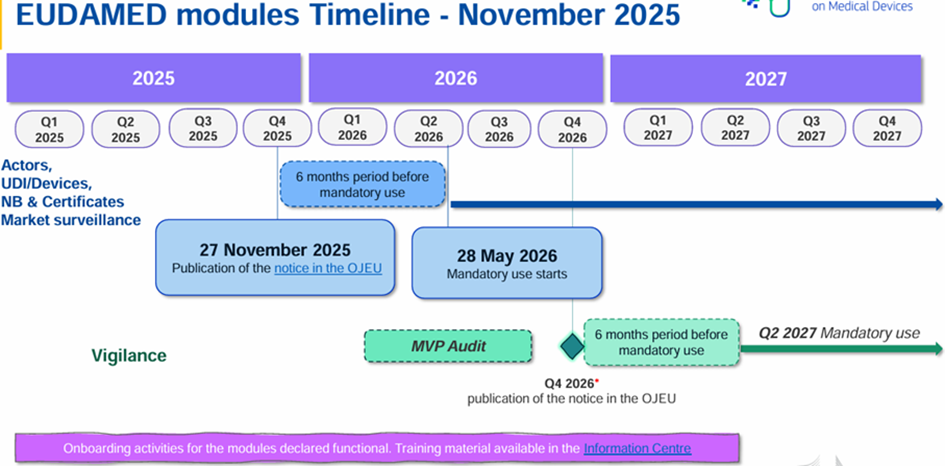
Some modules are already available and can be used voluntarily. A roadmap project was released on July 10, 2024, indicating a full deployment of EUDAMED scheduled for the second quarter of 2027.
On 27 November 2025, the European Commission published Decision (EU) 2025/2371 on the functionality and compliance with specifications for EUDAMED modules:
- Registration of economic operators” module (ACT module)
- Medical device registration and UDI database module (UDI/DEV module)
- Certificates and notified bodies module (NB/CRF module)
- Market surveillance module (MSU module)
These 4 modules are therefore operational and will be compulsory from 28 May 2026 (i.e. 6 months after publication in the OJEU) in accordance with Regulation (EU) 2024/1860.
The remaining 2 modules are not currently available:
- Clinical investigations/performance studies module (CI/PS module)
- Post-trade surveillance and vigilance module (VGL module)
A roadmap has been updated:
Useful Documents and Guides:
Q&A “on practical aspects related to the implementation of the gradual roll-out of Eudamed pursuant to the MDR and IVDR, as amended by Regulation (EU) 2024/1860 amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards a gradual roll-out of Eudamed, the obligation to inform in case of interruption or discontinuation of supply, and transitional provisions for certain in vitro diagnostic medical devices” (22/11/2024)
USER GUIDES to record the information in EUDAMED
MDCG “EUDAMED”
European Commission – The EUDAMED four first modules will be mandatory to use as from 28 May 2026
OJEU – Commission Decision (EU) 2025/2371 of 26 November 2025 concerning the opinion on the functionality and compliance with functional specifications of certain electronic systems included in the European database on medical devices referred to in Article 34(1) of Regulation (EU) 2017/745 of the European Parliament and of the Council
Article written by Camille Neermul