Implementation of ICH Q12 in Europe: where are we now?
What does the ICH Q12 guideline consist of?
The main purpose of the ICH Q12 “Guideline on technical and regulatory considerations for pharmaceutical product lifecycle management” is to provide a framework to facilitate the management of post-approval changes relating to Module 3 of the Marketing Authorisation (MA), or “CMC” changes, in a more predictable and efficient manner.
These guidelines and their appendices were adopted by the EMA’s Committee for Medicinal Products for Human Use (CHMP) in January 2020.
This text aims to promote innovation and continuous improvement, and to strengthen quality assurance and reliability of product supply, including proactive planning of adjustments to the supply chain in a context of transparency between the pharmaceutical industry and health authorities.
It also strives to promote, among regulators (evaluators and inspectors), a better understanding of applicants’ pharmaceutical quality management systems (QMS) for the management of post-marketing CMC changes, with a view to minimising regulatory variations in an international context.
Finally, it complements the ICH Q8 to ICH Q11 guidelines, offering opportunities for a science-based and risk-based approach to evaluating changes throughout the drug lifecycle:
- ICH Q8 “Pharmaceutical Development” / ICH Q11 “Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities)”: focus on the early stages of the product life cycle (development, registration, and launch);
- ICH Q9 “Quality Risk Management“ (QRM): describes the principles and tools for quality risk management that can be applied at different levels;
- ICH Q10 “Pharmaceutical Quality System“(PQS): describes an evolving model of a quality management system that can be implemented throughout the product lifecycle.
ICH Q12 aims to demonstrate how better knowledge of products and processes can help to determine more accurately which post-approval changes actually require regulatory submission.
To this end, the final version of the text adopted by the ICH in November 2019 presents four regulatory tools and four regulatory facilitators, accompanied by guiding principles, intended to harmonize the management of post-marketing CMC changes at the global level (see Table 1).
Table 1: ICH Q12 regulatory tools and facilitators
| Regulatory tools | Categorisation of Post-Approval CMC Changes | Facilitators | Pharmaceutical Quality System (PQS) and Change Management |
| Established Conditions (ECs) | Relationship Between Regulatory Assessment and Inspection | ||
| Post-Approval Change Management Protocol (PACMP) | Structured Approaches for Frequent CMC Post-Approval Changes | ||
| Product Lifecycle Management (PLCM) Document | Stability Data Approaches to Support the Evaluation of CMC Changes |
Towards easier implementation of the full potential offered by ICH Q12
In March 2020, the European Commission and the EMA published an explanatory note on the implementation with restrictions of this guideline in the European Union (i.e., ICH Q12 Step 5).
At the time of publication of this note, conceptual differences between the content of ICH Q12 and the EU legal framework were identified as potentially hindering the full implementation of this new guideline in Europe. These differences did not allow for the full application of the operational and regulatory flexibility defined in ICH Q12.
In particular, additional risk-based scientific approaches for defining Established Conditions (EC, see Chapter 3 of ICH Q12) and associated notification categories, as well as the Product Lifecycle Management Document (PLCM, see Chapter 5 of ICH Q12), were not considered compatible with the EU legal framework for variations* in force at that time.
Following the revision of the regulation on modifications applicable since January 1, 2025 in Europe, the European Commission adopted and published on September 22, 2025 the final version of the guidelines on the details of the different categories of modifications and the functioning of the procedures (applicable from January 15, 2026). This regulation is therefore now compatible with ICH Q12 for the subjects it concerns (see Table 2).
With regards to the Pharmaceutical Quality System (PQS, see Chapter 6 of ICH Q12), and change management, as well as the link between regulatory assessment and inspection (see Chapter 7 of ICH Q12), some additional clarifications regarding the demonstration and evaluation of the effectiveness of the QMS and communication between regulatory authorities still require further consideration during the implementation of the guideline (see Table 2).
Table 2: European regulatory developments versus key ICH Q12 tools/concepts
| ICHQ12 – Key tools/concepts | Variations guidelines (2013) | Variations guidelines (2025) | Comments |
| Chapter 2 – Categorisation of Post-Approval CMC Changes | 2025 Guidelines: Addition of additional ICH Q12-compliant regulatory tools (QbD, PACMP, PLCM) in cat. Q.I.e & Q.II.g | ||
| Chapter 3 – Established Conditions (ECs) | ECs generally reflect information and quality characteristics that are subject to variation. | ||
| Chapter 4 – Post-Approval Change Management Protocol (PACMP) | 2025 Guidelines: > Active substance: cat. Q.I.e.2 to Q.I.e.5 > Finished product: cat. Q.II.g.2 to Q.II.g.5 | ||
| Chapter 5 – Product Lifecycle Management (PLCM) Document | 2025 Guidelines: > Active substance: cat. Q.I.e.6 to Q.I.e.8 > Finished product: cat. Q.II.g.6 to Q.II.g.8 | ||
| Chapter 6 – Pharmaceutical Quality System (PQS) and Change Management | – | – | See ICH Q10 (PQS) & GMP (chapter 1) – clarifications expected |
| Chapter 7 – Relationship Between Regulatory Assessment and Inspection | – | – | clarifications expected |
| Chapter 8 – Structured Approaches for Frequent CMC Post-Approval Changes | – | – | clarifications expected |
| Chapter 9 – Stability Data Approaches to Support the Evaluation of CMC Changes | – | – | See draft ICH Q1 – clarifications expected |
Compatible //
Not compatible // – No reference
To summarise, changes in European regulations are effectively tending to make the implementation of ICH Q12 necessary in order to benefit from all the advances envisaged in this text and thus facilitate the management of CMC changes after marketing authorization in a more predictable and efficient manner.
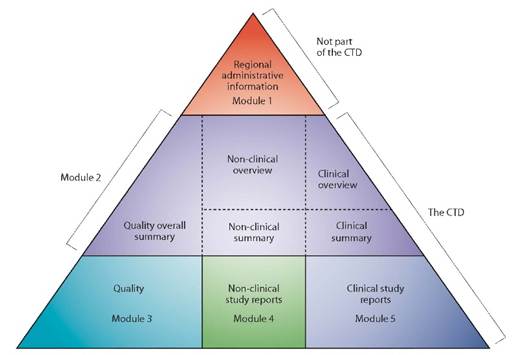
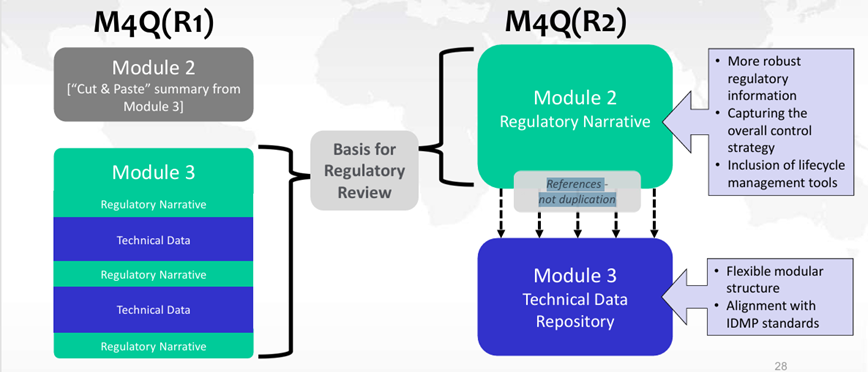
Furthermore, a future revision of the ICH Q12 guidelines is to be expected once the ICH M4Q(R2) “CTD on Quality” guideline has been adopted by the CHMP in Europe, in particular to amend Appendix 1 (relating to the sections of the CTD containing ECs).
* EC Variations Guidelines (2013) = Guidelines on the details of the various categories of variations, on the operation of the procedures laid down in Chapters II, IIa, III and IV of Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products and on the documentation to be submitted pursuant to those procedures (2013/C 223/01)
** EC Variations Guidelines (2025)= Guidelines on the details of the various categories of variations, on the operation of the procedures laid down in Chapters II, IIa, III and IV of Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products and on the documentation to be submitted pursuant to those procedures (C/2025/5045)
Sources:
– ICH – Quality Guidelines
– EMA – Guidance on the application of the revised variations framework
Article written by par Isabelle MOUVAULT, Senior Advisor in Pharmaceutical Affairs