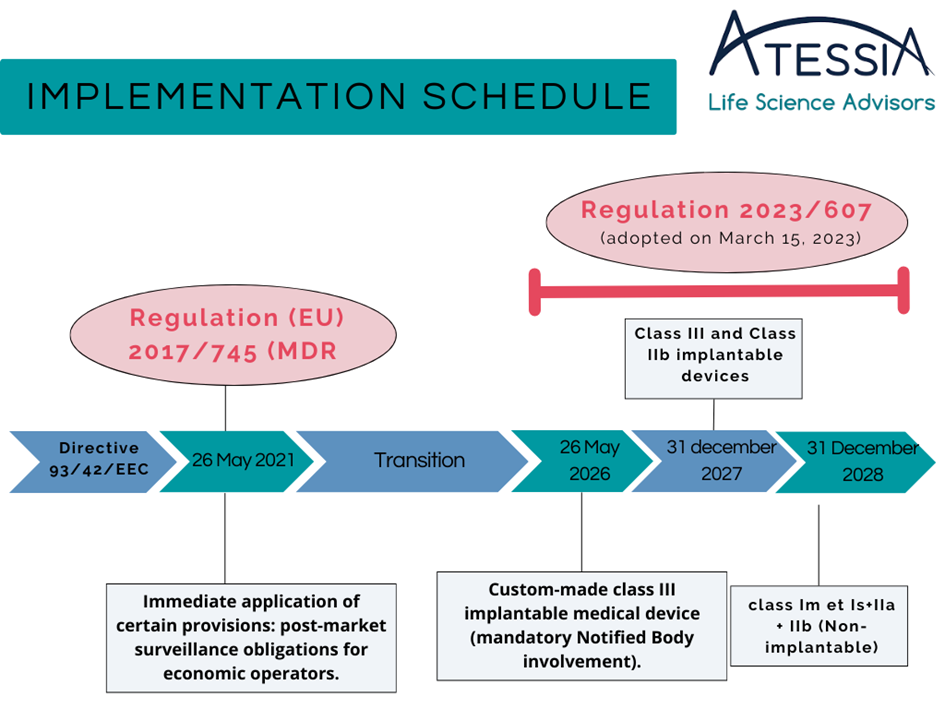
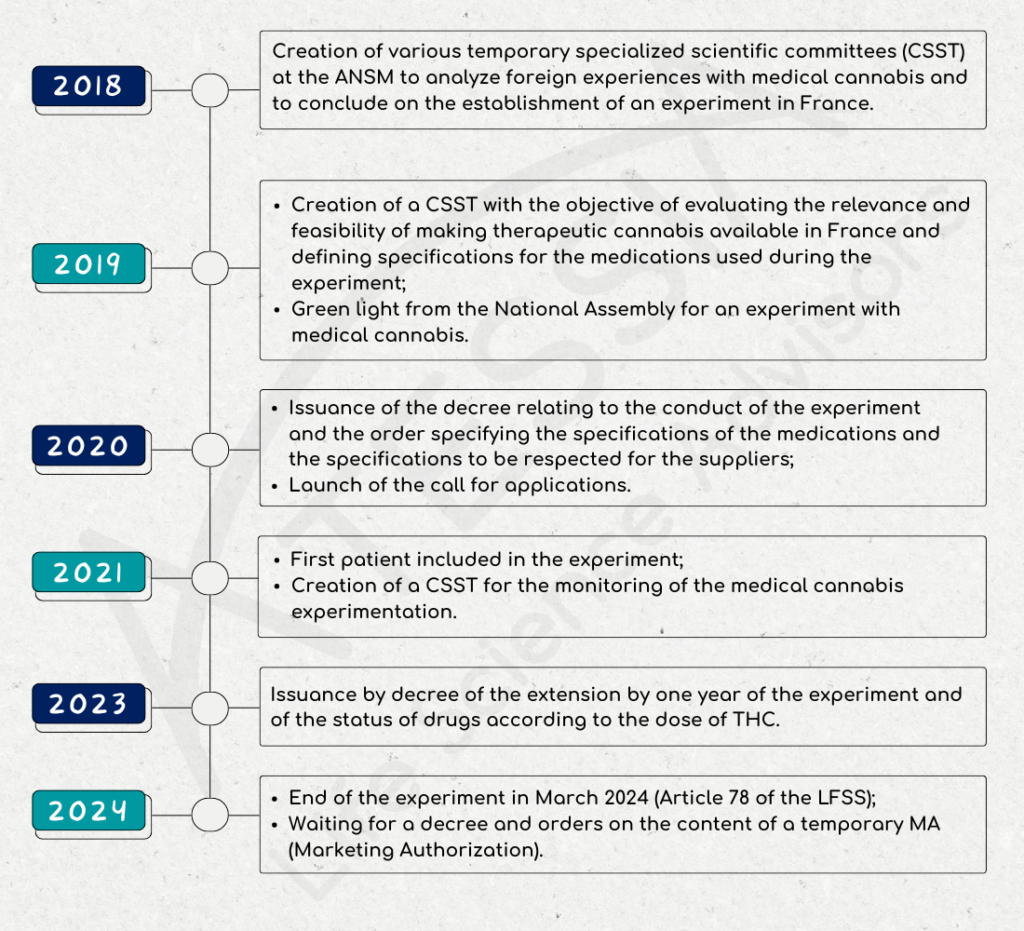
Forbes – ATESSIA, Life Science Advisors : The Regulatory and Pharmaceutical Affairs Consulting Firm Dedicated to the Healthcare Industry
Leveraging its expertise, ATESSIA supports a wide range of health products, with a particular focus on pharmaceuticals and medical devices. Through a tailored and rigorous approach, the firm helps its clients navigate complex regulatory landscapes, ensuring compliance and innovation in their projects.
More Human, More Agile
“I founded ATESSIA, Life Science Advisors, with the conviction that regulatory and pharmaceutical affairs consulting could be reimagined through a more human and agile lens,” explains Géraldine Baudot-Visser. Drawing on several years of experience in the pharmaceutical industry and renowned consulting firms, I recognized the opportunity to create a company that prioritizes client satisfaction and the empowerment of its experts while ensuring unwavering excellence in the pharmaceutical field.” The initial challenges naturally revolved around gaining the trust of the first clients and dismantling stereotypes about consulting. The firm needed to prove that rigor and creativity can coexist and demonstrate that a consulting firm can combine cutting-edge expertise with a bespoke approach. Striking the balance between managing a young company and delivering excellence has been a top priority since day one. For the founder, the goal was clear: to become a reference in regulatory consulting in the French and European markets within a few years, despite competition from established firms.
Tailor-Made Solutions, Continuously Enhanced
Every phase of ATESSIA’s growth has been driven by the ambition to establish itself as a leader while rallying employees around its clients’ challenges. Each step has also been an opportunity to reinvent, innovate, and grow while staying true to its values to ensure a client-centered approach. “For example,” says the founder, “our regulatory intelligence service is essential for our pharmaceutical clients, achieving a satisfaction and retention rate exceeding 90%. This success reflects our ability to provide tailored support and anticipate market changes, meeting the most demanding expectations of our partners.” This commitment to exceeding expectations relies on a team capable of anticipating, adapting, and ensuring compliance with the strictest standards. This collective dynamic keeps ATESSIA at the forefront, offering clients ever more efficient, custom-tailored solutions.
An Excellence-Driven Approach
“Our clients choose us because they seek far more than standardized solutions. At ATESSIA, we work with the precision of master artisans, designing bespoke solutions perfectly adapted to the specificities of each project in a highly regulated environment. Like the finest craftsmen of traditional expertise, we pay particular attention to ensuring impeccable quality in our services to provide reliable technical support to our clients,” shares Géraldine Baudot-Visser. This excellence-driven approach makes ATESSIA a trusted partner for its clients. The consulting firm builds strong relationships founded on attentive listening, in-depth expertise, and the ability to anticipate market changes. This proactive, personalized vision allows ATESSIA to deliver solutions that are always perfectly adapted and tailored.
“Every contribution we make to the healthcare industry, no matter how modest, has the potential to impact someone’s life somewhere. This gives profound meaning to what we do and makes every project-whether centered on pharmaceutical facilities, system quality, or innovation-much more than just a mission for us: it’s an opportunity to actively contribute to improving patients’ lives. This responsibility is a source of immense and invaluable pride,” she concludes.
https://www.forbes.fr/mediasfrance/atessia/