Clinical Trials versus Research Involving Human Subjects (RIPH): How Are Pharmaceutical Companies Affected?
Medical research constitutes an essential lever for the development of new treatments and for the continuous improvement of patient care.
In France, this research is governed by strict regulations designed to protect participants and ensure the integrity of human subjects. The Jardé Law, adopted in March 2012 and fully applied since November 2016, constitutes the legal framework for research involving human subjects (RIPH).
Relationship between the Jardé Law and the European Clinical Trials Framework
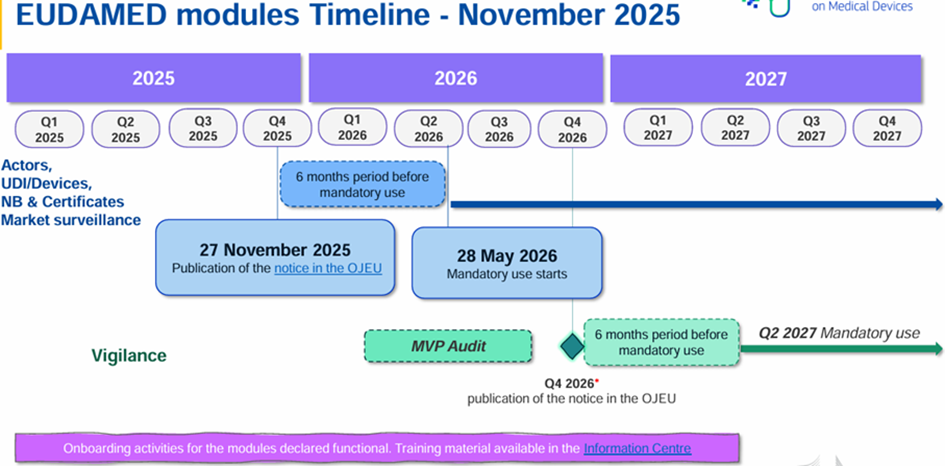
It should be noted that clinical trials involving medicinal products are primarily regulated by the EU Clinical Trials Regulation (EU) 536/2014 (CTR), which came into effect on January 31, 2022. This regulation repealed and replaced Directive 2001/20/EC. Since that date, clinical trials on drugs must be submitted via the Clinical Trials Information System (CTIS). As a reminder, any clinical trial with at least one active investigator site in France as of January 31, 2025 had to undergo a transition request to CTIS by its sponsor. The ANSM notice to sponsors provides practical guidance on the submission of applications.
The Jardé Law does not replace the CTR but complements the European framework with specific national requirements, notably regarding the protection of participants, consent and information, research classification, and procedures involving the Committees for the Protection of Persons (CPP).
Other regulatory provisions must also be integrated by sponsors, such as procedures on personal data protection (GDPR, CNIL rfefernce methodologies), procedures related to the use of medicinal products composed wholly or partially of genetically modified organisms (GMOs), and specific rules applicable to certain health products.
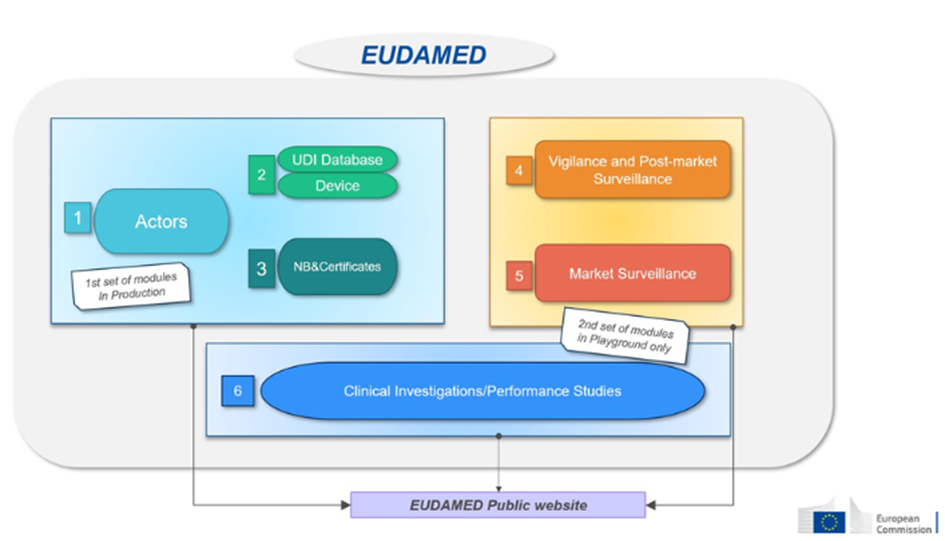
Indeed, depending on the nature of the research object, the applicable provisions differ. The Jardé Law thus covers: drugs, medical devices (clinical investigation) in vitro diagnostic medical devices (performance study), cell therapy products, tissues, organs, labile blood products (LBPs), or even research on dietary supplements or cosmetics.
What is the Jardé Law?
The Jardé Law, named after Deputy Olivier Jardé, is a regulation that governs the conditions under which research involving human participants can be conducted in France. It replaces the Huriet-Sérusclat Law of 1988 and aims to strengthen the protection of participants while simplifying and harmonizing procedures, taking into account the level of risk incurred.
The main reference texts include:
– The Jardé Law, Law No. 2012-300 of March 5, 2012, relating to research involving human subjects.
– The ordinance, known as the “modified Jardé Law,” relating to research involving human subjects.
– Decree No. 2016-1537 of November 16, 2016, relating to research involving human subjects.
Classification of RIPH
Research organized and conducted on human beings with the aim of developing biological or medical knowledge is referred to as “research involving human subjects” (RIPH). There are three types of RIPH:
| Category | Legal Provisions | Framework |
| Category 1 Interventional research involving a risk to participants (intervention not justified by usual care) | Articles L1121-1 to L1121-17 of the Public Health Code | These studies require prior authorization from the ANSM (French National Agency for Medicines and Health Products Safety) and a favorable opinion from a Committee for the Protection of Persons (CPP). |
| Category 2: Interventional research with minimal risks and constraints * | Articles L1121-1 to L1121-17 of the Public Health Code | These studies require a favorable opinion from a CPP, but not authorization from the ANSM. |
| Category 3: Non-interventional research (observational studies – negligible risks) | Article L1121-1 of Public Health Code | These studies require a favorable opinion from a CPP, but not authorization from the ANSM. |
* Research involving drugs cannot in principle fall under Category 2, except for very specific cases defined by regulation. An order sets the criteria to be met to remain within the scope of RIPH 2.
What Are the Implications for the Industry?
Participant Information and consent
The objective of the Jardé Law is to ensure the safety of participants. Special attention is given to the notions of informed consent and clear, understandable, and fair information.
Industrials, as sponsors, must ensure that participants fully understand the stakes, procedures, risks and constraints, and potential benefits of the study. These requirements are detailed in Articles L1122-1-1 to L1122-2 of the Public Health Code (CSP).
Determination of Competent Authorities and Applicable Procedures
Sponsors must determine the category of their research during the design phase, ensure they obtain the necessary authorizations (CPP and/or ANSM), and define the applicable procedure (CTR/CTIS for drugs).
Interactions with CPPs and the ANSM
CPPs are French ethics committees (Committees for the Protection of Persons) responsible for evaluating participant protection, the quality of information and consent, and the benefit/risk balance. Interactions with CPPs and the ANSM are essential for the validation of research projects. Good communication and submission of complete dossiers are necessary, in accordance with Articles L1123-6 and L1123-7 of the Public Health Code. For each RIPH, a CPP responsible for assessing the application is selected by random draw. The information system for research involving the human person (SI RIPH 2G) enables the submission of an application for an opinion and the random designation of a CPP. The platform also allows for the declaration of healthy volunteers participating in a study.
Procedures
Before submitting the authorization request dossier (initial authorization and substantial modification) and/or human research opinion request, or routine care research, sponsors must obtain an IDRCB registration number for the research. This number identifies each research conducted in France. For a interventional research authorization and opinion request concerning a medicinal product for human use, sponsors must instead obtain a research registration number in the European CTIS database (formerly: EudraCT).
Subsequently, sponsors will electronically submit the biomedical research authorization and/or opinion request dossier to the ANSM and/or CPP, in accordance with the current orders setting the dossier formats for each type of research. Various “Notices to Sponsors” guide these procedures according to the situation.
Conclusion
The Jardé Law thus ensures the safety of participants in clinical research in France.
For health manufacturers, understanding and complying with these regulations is not only a legal obligation but also a guarantee of the quality of the data generated, particularly for use in a Marketing Authorization Application (MAA) dossier.
By integrating the requirements of the Jardé Law into their processes, manufacturers contribute to the development of innovative treatments while ensuring high ethical standards, in accordance with French and European regulations on this subject.
Atessia supports you in implementing these processes with its expertise in clinical trials.
Article written by Mathilde ISRAEL