

Comprendre EUDAMED : La Base de Données Européenne sur les Dispositifs Médicaux
Le règlement (UE) 2017/745 introduit de nouvelles exigences afin de renforcer la sécurité des patients et utilisateurs. Une des nouveautés de ce nouveau règlement est la création d’une base de données européenne dédiée aux informations sur les dispositifs médicaux appelée EUDAMED.
Cette plateforme sécurisée permettra :
- d’accroître la transparence des informations sur les dispositifs médicaux avec un accès au grand public
- une meilleure coordination entre les États membres dans la surveillance post commercialisation des dispositifs médicaux
EUDAMED est une plateforme sécurisée utilisée pour recueillir et partager des données relatives aux dispositifs médicaux mis sur le marché de l’Union européenne ainsi que ceux faisant l’objet d’investigation clinique.
Le règlement introduit de nouvelles exigences applicables aux différents acteurs pour EUDAMED.
Cette base de données sera composée de 6 modules connectés les uns aux autres :
| Module : | Qui doit enregistrer des informations ? | Accessible au public | |
| 1-Acteurs | Les opérateurs économiques doivent s’enregistrer en tant qu’acteur dans EUDAMED et fournir les informations requises. | -Fabricants de l’UE et de pays tiers, -Mandataires, -Producteurs d’emballages de systèmes/procédures – Importateurs | Disponible sur base volontaire depuis décembre 2020 et sera obligatoire à partir de T4 2027 |
| 2-Dispositifs | Les fabricants doivent soumettre dans EUDAMED le basic-IUD et les informations de tous les dispositifs qu’ils mettent sur le marché de l’UE. | Uniquement les fabricants Enregistrement des dispositifs médicaux sous MDR Aucune obligation pour les legacy devices (si enregistrement dans EUDAMED, il faudra faire un nouvel enregistrement pour les produits sous MDR, considérés comme de nouveaux produits) | Disponible sur base volontaire depuis octobre 2021 et est obligatoire à partir de T1 2026 |
| 3-Organismes notifiés (ON) et certificats | Les organismes notifiés (ON) doivent enregistrer dans EUDAMED toute information concernant les certificats délivrés, suspendus, rétablis, retirés ou refusés et les autres restrictions imposées à ces certificats. Ces informations sont accessibles au public. | Organismes Notifiés | Disponible sur base volontaire depuis octobre 2021 et est obligatoire à partir de T1 2026 |
| 4-Vigilance | Module dédié à tous les rapports de vigilance et de surveillance post-commercialisation. -information de sécurité (Field Safety notice, FSN) -Actions correctives de sécurité (Field Safety Corrective Action, FSCA) -Rapport d’investigation des causes d’incident et mesures correctives (MIR) -Rapport de tendances (trend report) -Rapport périodique de sécurité (PSUR) | Fabricants | Sera obligatoire à partir de T4 2027 |
| 5-Surveillance du marché | La coordination des actions de surveillance de marché entre les différentes autorités compétentes. | Autorités compétentes uniquement | Sera obligatoire à partir de T4 2027 |
| 6-CI/PS : (Clinical investigation/ Performance studies) : | Ce module concerne les enregistrements des investigations cliniques (DM) et études de performance (DMDIV). Rapport et résumé d’investigation clinique Evènement indésirable grave survenu pendant les investigations cliniques | Promoteurs | Sera obligatoire à partir de T4 2027 |
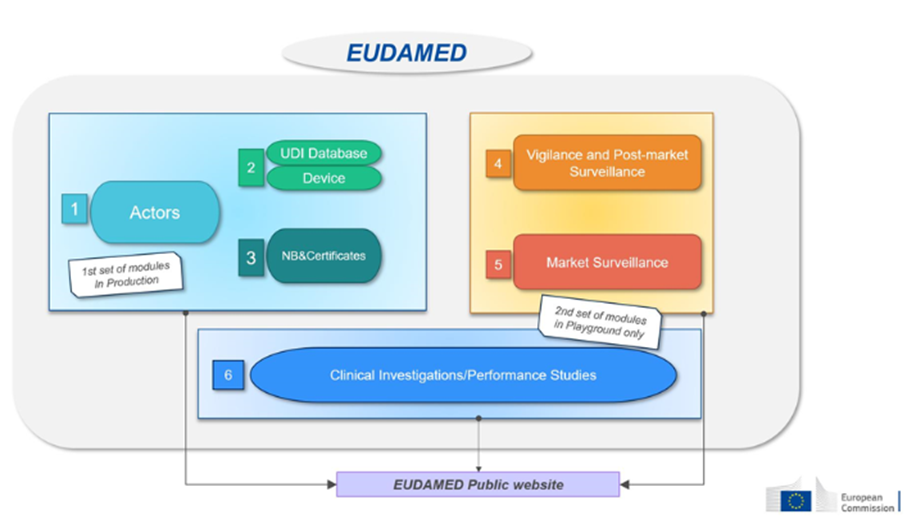
Source : Commission européenne
Et les distributeurs ?
Le MDR n’impose aucune exigence aux distributeurs concernant EUDAMED. Ils n’ont donc aucun accès sécurisé dans EUDAMED et ils ont uniquement l’accès grand public. Certains pays peuvent cependant définir des exigences supplémentaires, c’est le cas de la France qui demande aux distributeurs de s’enregistrer via le formulaire ANSM.
Calendrier de déploiement EUDAMED
En octobre 2019, la Commission européenne avait annoncé le report du lancement d’EUDAMED de 2 ans à mai 2022.
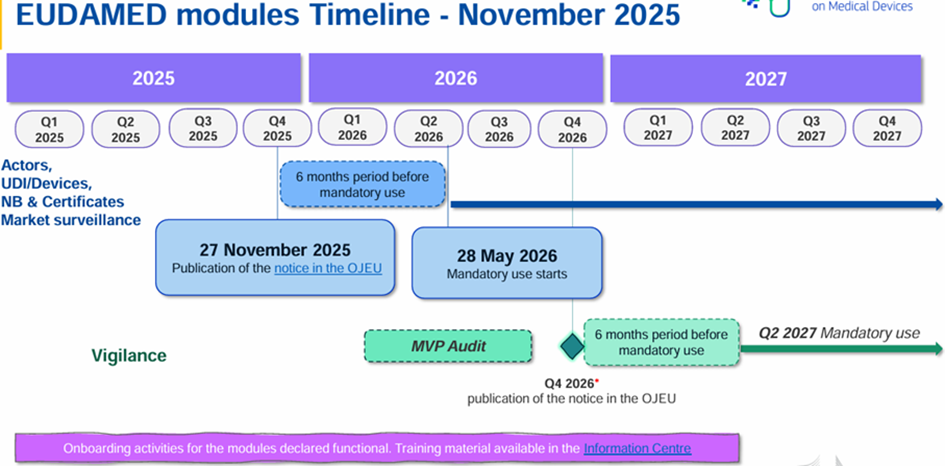
Certains modules sont déjà disponibles et peuvent être utilisés volontairement. Un projet de roadmap est sorti le 10 juillet 2024 indiquant un déploiement total d’EUDAMED prévu au deuxième trimestre 2027.
La Commission européenne a publié ce 27 novembre 2025 une décision (UE) 2025/2371 concernant la fonctionnalité et le respect des spécifications pour les modules EUDAMED :
- Module « enregistrement des opérateurs économiques » (ACT module)
- Module « enregistrement des dispositifs médicaux et base de données UDI » (UDI/DEV module)
- Module « certificats et organismes notifiés » (NB/CRF module)
- Module « surveillance sur le marché » (MSU module)
Ainsi ces 4 modules sont donc opérationnels et ils seront obligatoires à partir du 28 mai 2026 (soit 6 mois après la publication au JOUE) selon le règlement (UE) 2024/1860.
Pour les 2 modules restants, ils ne sont pour le moment pas disponibles :
- Module investigations cliniques/studes de performance (CI/PS module)
- Module surveillance post-marché et vigilance (VGL module)
Une roadmap a été mise à jour :
Documents et guides utiles :
- Q&A “on practical aspects related to the implementation of the gradual roll-out of Eudamed pursuant to the MDR and IVDR, as amended by Regulation (EU) 2024/1860 amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards a gradual roll-out of Eudamed, the obligation to inform in case of interruption or discontinuation of supply, and transitional provisions for certain in vitro diagnostic medical devices” (22/11/2024)
- USER GUIDES afin d’enregistrer les informations dans EUDAMED
- MDCG “EUDAMED”
- Commission européenne – The EUDAMED four first modules will be mandatory to use as from 28 May 2026 (27/11/2025)
- JOUE – Décision (UE) 2025/2371 de la Commission du 26 novembre 2025 relative à l’avis concernant la fonctionnalité et le respect des spécifications fonctionnelles de certains systèmes électroniques figurant dans la base de données européenne sur les dispositifs médicaux visée à l’article 34, paragraphe 1, du règlement (UE) 2017/745 du Parlement européen et du Conseil (27/11/2025)
