

Background
In Europe, information relating to the quality, safety, and efficacy of the Marketing Authorization Application (MAA) for medicinal products is compiled in a common format known as the Common Technical Document (CTD) format. The CTD format applies in all regions that recognize ICH guidelines. It is currently organized into five Modules: Module 1 is specific to the region, while Modules 2, 3, 4, and 5 are common to all regions (see Figure 1).
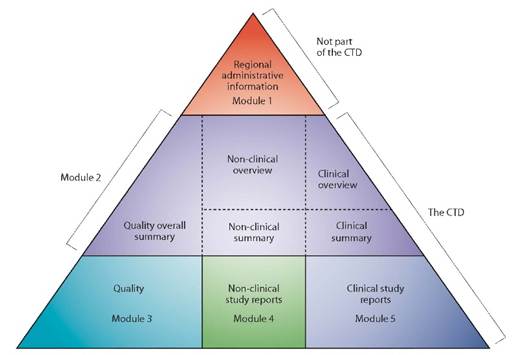
Figure 1 : The CTD triangle
The CTD format, described in ICH M4, became the mandatory format for marketing authorization applications for new drugs in Europe in July 2003. Prior to its implementation, marketing authorization dossiers met the requirements of the NtA Volume 2B format (1998 edition).
For industries, the introduction of the CTD format eliminated the need to reformat information submitted to the various ICH regulatory authorities.
Subsequently, the introduction of the ‘eCTD’ (electronic Common Technical Document) format, which first became mandatory in Europe in 2007 for centralized marketing authorizations, revolutionized regulatory practices by harmonizing electronic submissions to ICH regulatory authorities, notably replacing submissions in NeeS (Non-eCTD electronic Submission) format. Information on the eCTD format is available in ICH M8.
Reasons for the revision of ICH M4Q(R1)
In Europe, the content of Module 2.3 (QOS – Quality Overall Summary) and Module 3 of the marketing authorisation dossier meets the requirements detailed in the ICH M4Q(R1) guidelines implemented in July 2003. No revision of this text has been carried out for more than 20 years, which has led legislators to several observations:
Observation #1) The traditional CTD structure is not suited to addressing modern quality concepts.
Since the publication of ICH M4Q(R1), new guidelines ICH Q8 to ICH Q14 have been developed, introducing innovative concepts such as Quality by Design (QbD), Quality Risk Management (QRM) and Life Cycle Management (LCM) approaches, as well as Continuous Manufacturing (CM).
The ICH M4Q(R1) document was not designed to take into account the new quality principles, and integrating them into the current CTD format is not straightforward.
Observation #2) The traditional CTD structure is not suited to handling changes in technologies and product types.
ICH M4Q(R1) was designed primarily for conventional small molecules and is structured around the active substance (Part 32S) and the finished product (Part 32P), with adaptations for biological products. Experience has shown that complex products and new therapeutic modalities (nanomedicines, oligonucleotides and biological products such as vaccines, cell and gene therapies, and tissue-engineered products) as well as combination products often do not fit perfectly into this framework.
Observation #3) The traditional CTD structure creates ambiguity in the organisation and placement of information.
The required modular format (i.e. summary of quality data included in Module 2.3 and detailed information included in Module 3) leaves room for differing interpretations as to which details should be included in Module 2.3 versus Module 3 and often leads to repetition of information. Ambiguity remains regarding the location of information and cross-references between modules.
The management of updates and variations to marketing authorizations throughout the lifecycle, while maintaining consistency in the CTD structure with ICH M4Q(R1), is not optimal.
Observation #4) The traditional CTD structure allows regional differences to remain.
Despite the harmonization of the dossier format within the ICH, additional requirements specific to certain countries/regions often persist, thereby reducing the advantage of having a single format.
Observation #5) The current eCTD format does not allow for the integration of new requirements for electronic and structured data.
The current trend is towards structured, machine-readable submissions and the use of data standards (e.g. ISO IDMP 11615), in line with the future implementation of SPQS (Structured Product Quality Submission) by the EMA, which will lead to future ICH M16 guidelines (see Figure 2).
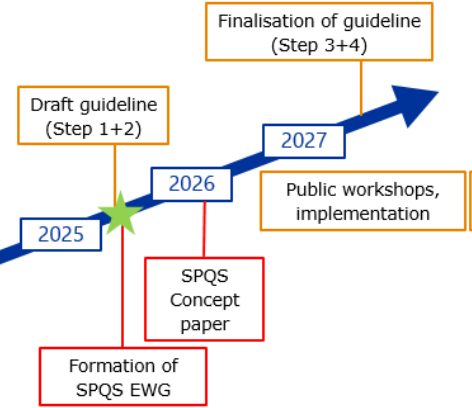
Figure 2: Interactions between ICH M4Q(R2) and SPQS (future ICH M16)
ICH M4Q(R1) was not designed for such structured content, which complicates automation and prevents data reuse between submissions.
For all these reasons, ICH MQ4(R1) must be redesigned to enable data management and standardisation, thereby promoting the efficiency of the dossier review and approval process.
Overview of the ICH M4Q(R1) revision: current framework versus future framework
Although changes are being made to the location of information in the future Modules 2.3 and 3, these do not alter regulatory expectations. The data used as the basis for regulatory assessment, previously presented in Module 3, will now be included in Module 2.3. Module 3 will now serve as a repository for technical data (protocols, reports, data, etc.) (see Figure 3).
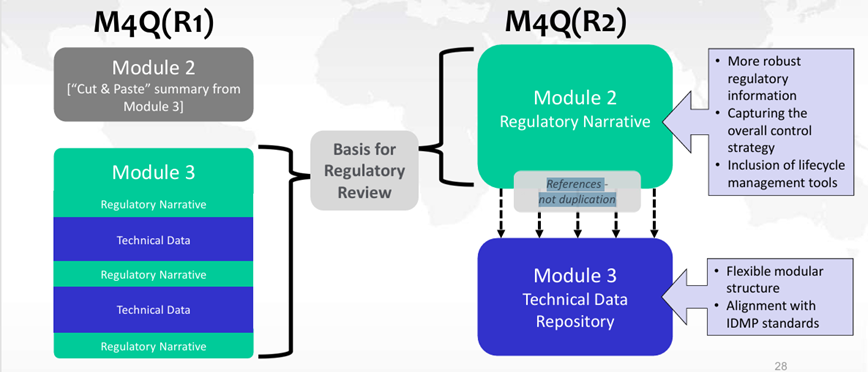
Figure 3: Changes in data location between ICH M4Q(R1) and ICH M4Q(R2)
The future granularity of Module 2.3 and Module 3, as anticipated in the 14 May 2025 draft version of ICH M4Q(R2), is presented in Figure 4 and Figure 5.

Figure 4: Granularity of Module 2.3 according to ICH M4Q(R2) (1)

Figure 4: Granularity of Module 2.3 according to ICH M4Q(R2) (2)

Figure 4: Granularity of Module 2.3 according to ICH M4Q(R2) (3)

Figure 5: Granularity of Module 3 according to ICH M4Q(R2)
Essential information on quality (2.3.3 Core Quality Information (CQI)):
– shall include all information subject to lifecycle management in accordance with regional requirements for post-authorisation changes to ensure product quality.
– shall be maintained throughout the product lifecycle to ensure that product quality information remains current.
The information contained in sections 2.3.1 (General Information), 2.3.2 (Overall Development and Overall Control Strategy), 2.3.4 (Development Summary and Justification), 2.3.5 (Product Lifecycle Management), 2.3.6 (Product Quality Benefit Risk) and Module 3 will be supportive information, which may be modified or supplemented for post-marketing submissions.
In the future ICH M4Q(R2), information will be grouped into specific subsections of materials/components used in manufacturing:
- drug substances (DS),
- substances intermediates (SI),
- raw materials (RM),
- starting/source materials (SM),
- excipients (EX),
- reference materials (RS),
- impurities (IM),
- drug products (DP),
- products intermediates (PI),
- packaged medicinal products (PM),
- pharmaceutical products (PH) and
- medical devices (MD).
Information relating to analytical procedures and facilities that apply to all materials will be presented in dedicated sections.
Each subsection will then be organized according to the following DMCS structure:
- Description: identifies the material and its key characteristics;
- Manufacture: outlines the production process and process controls;
- Control: Control: describes quality control measures such as specifications;
- Storage: provides stability, container closure information, and retest period/shelf-life
The relationships between Module 2.3 and Module 3 in the context of the DMCS model used for materials/components are illustrated as follows:
| 2.3.3 Core Quality Information | 2.3.4 Development Summary and Justification | 3.2 Body of Data | |
| Information related to what the material is and its key characteristics, which is considered necessary to enable marketing authorization and facilitate lifecycle management | Scientific and risk-based development summary and justifications related to what the material is and its key characteristics | Supportive information including reports and data related to what the material is and its key characteristics | |
| Description | Nomenclature, structure, composition, key characteristics | Characterization summary, formulation development and justification | Characterization data, formulation development and justification data |
| Manufacture | Manufacturing process description, IPCs, critical process parameters | Process development and validation/evaluation summary | Process development and validation/evaluation data |
| Control | Specifications | Overview of batch analysis, justification of specifications | Batch analysis and justification data |
| Storage | Container closure system description, storage conditions, and retest period/shelf life | Overview of stability studies, justification of proposed container closure system | Container closure selection and stability data |
Conclusion
In conclusion, ICH M4Q(R2) aims to promote harmonization of the quality content of dossiers, ideally enabling a single dossier to be submitted in all ICH member countries.
Where required by law, the applicant shall provide any additional region-specific information directly in the relevant section of a separate document, in the form of an addendum to the harmonized base document used in all ICH regions.
Atessia supports pharmaceutical companies in preparing Modules 2.3 and 3.
Sources:
– ICH M4 Organisation Including the Granularity document that provides guidance on document location and pagination
– Notice to Applicants, Volume 2B incorporating the Common Technical Document (CTD) (May 2008)
– ICH M4Q(R1) CTD on Quality
– ICH M4Q Q&As (R1) Questions & Answers: CTD on Quality
– ICH M4Q(R2) EWG Revision of M4Q(R1) (draft guideline 14 May 2025)
– M4Q(R2) Step 2 presentation (18 June 2025)
– Présentation Finding your way with the new eCTD (ICH-M4Q), Ivica Malnar, Agency for Medicinal Products and Medical Devices (HALMED) (23-24 September 2025)
– ICH M8 electronic Common Technical Document (eCTD) v3.2.2
– ICH M8 electronic Common Technical Document (eCTD) v4.0
Article written by Isabelle MOUVAULT, Senior Advisor in Pharmaceutical Affairs